Human Hyaluronidase in Disease Research
Cancer, Inflammation, Tissue Remodeling, and Drug Penetration
Abstract
Human hyaluronidases (HYALs) are a family of enzymes that catalyze the degradation of hyaluronan (HA), a major glycosaminoglycan component of the extracellular matrix (ECM). Aberrant hyaluronidase activity—whether elevated or suppressed—has been implicated in a broad spectrum of pathological conditions, including cancer progression, chronic inflammation, tissue fibrosis, and impaired drug delivery. This literature review synthesizes current evidence on the mechanistic roles of human hyaluronidase in disease microenvironments, with particular emphasis on matrix remodeling in solid tumors, immune cell trafficking, wound healing dynamics, and the emerging therapeutic strategy of enzymatic ECM modulation to enhance drug penetration. We also address critical limitations in translating preclinical findings to clinical outcomes and propose future research directions.
human hyaluronidase disease research, hyaluronidase cancer, ECM remodeling, inflammation, hyaluronan accumulation, drug penetration, tissue fibrosis
1. Introduction: Why Hyaluronidase Is Central to Disease Research
Hyaluronan (HA) is a non-sulfated glycosaminoglycan that constitutes a major structural and signaling component of the extracellular matrix. Its concentration and molecular weight are tightly regulated by the balance between HA synthases (HAS1-3) and hyaluronidases (HYAL1-4, SPAM1, HYALP1). In healthy tissues, this equilibrium maintains tissue hydration, lubrication, and structural integrity. However, in disease states, dysregulation of this balance leads to either excessive HA accumulation or aberrant fragmentation—both of which profoundly alter tissue microenvironments.
Human hyaluronidases are endo-β-N-acetylhexosaminidases that cleave HA into smaller oligosaccharides. HYAL1 and HYAL2 are the primary somatic isoforms, with HYAL1 active in acidic lysosomal compartments and HYAL2 operating at the cell surface in concert with CD44. Genetic deficiencies in these enzymes result in lysosomal storage disorders (mucopolysaccharidosis type IX), whereas their overexpression or altered activity has been documented in multiple malignancies and inflammatory conditions. Consequently, recombinant human hyaluronidase has emerged as both a research tool and a therapeutic candidate for modulating ECM barriers in disease.
Hyaluronidase activity is a double-edged sword: insufficient degradation leads to HA accumulation and matrix densification, while excessive activity generates pro-inflammatory HA fragments that drive cell migration and angiogenesis.
2. Hyaluronan Accumulation and Tissue Microenvironment
HA accumulation is a hallmark of multiple disease microenvironments. In solid tumors, elevated HA deposition—driven by cancer-associated fibroblasts (CAFs) and tumor cells themselves—creates a hydrated, gel-like matrix that increases interstitial fluid pressure (IFP) and physically impedes drug diffusion. The accumulation of HA into the stroma varies by tumor type; malignant melanomas and squamous cell carcinomas typically exhibit low HA contents, whereas pancreatic ductal adenocarcinoma (PDAC) and certain breast cancers are characterized by dense HA-rich desmoplasia.
Beyond cancer, HA accumulation occurs in fibrotic organs, atherosclerotic plaques, and chronically inflamed joints. In liver fibrosis, for example, sustained HA deposition contributes to inflammation, tissue remodeling, and disease progression. Murine models of HYAL2 deficiency demonstrate that loss of hyaluronidase activity results in extracellular HA accumulation, leading to severe cardiopulmonary dysfunction characterized by expanded, disorganized valve leaflets and interstitial GAG deposition in the myocardium. These findings underscore that hyaluronidase-mediated HA turnover is essential for maintaining ECM homeostasis across organ systems.
Table 1. Tissue Microenvironment Changes Associated with HA Accumulation
| Disease Context | HA Source | Microenvironmental Effect | Clinical Consequence |
|---|---|---|---|
| Pancreatic cancer (PDAC) | CAFs, tumor cells | Elevated IFP; vascular compression | Poor chemotherapy perfusion |
| Liver fibrosis | Hepatic stellate cells | Matrix densification; inflammation | Progressive cirrhosis |
| Rheumatoid arthritis | Synovial fibroblasts | Joint space narrowing; immune cell retention | Cartilage degradation |
| Idiopathic pulmonary fibrosis | Alveolar epithelial cells | Stiffened parenchyma; impaired gas exchange | Respiratory failure |
| Atherosclerosis | Vascular smooth muscle cells | Plaque expansion; endothelial dysfunction | Myocardial infarction risk |
3. Cancer Research: Matrix Remodeling, Invasion, and Drug Penetration
3.1 Matrix Remodeling and Tumor Invasion
In the tumor microenvironment (TME), HA is intimately involved in cancer biology, modulating intracellular signaling pathways, cell proliferation, motility, and invasive properties. High molecular weight HA (HMW-HA) can create a favorable niche for tumor angiogenesis, invasion, and metastasis. Paradoxically, hyaluronidase-generated HA fragments also promote cancer progression by stimulating toll-like receptor (TLR) signaling and driving epithelial-mesenchymal transition (EMT).
Studies on biliary tract malignancies and pancreatic cancer have shown that aberrant hyaluronidase expression—both canonical (HYAL1/2) and non-canonical (TMEM2, CEMIP)—is frequently observed in solid tumors. This dual role creates a complex regulatory landscape where both HA accumulation and excessive HA turnover can promote malignancy. PEGPH20, a pegylated recombinant human hyaluronidase, has been extensively investigated as a strategy to degrade tumor HA, reduce stromal density, and improve vascular perfusion.
3.2 Drug Penetration Enhancement
HA-rich stroma presents a formidable barrier to nanomedicine and conventional chemotherapy. Intratumoral injection of hyaluronidase has been shown to decompose the ECM and decrease IFP in a concentration-dependent manner. Preclinical studies demonstrate that hyaluronidase-coated nanoparticles achieve deeper tumor penetration and enhanced cellular uptake compared to unmodified carriers. In HepG2 xenograft models, hyaluronidase-combined epirubicin-loaded nanoparticles (NPs-EPI/HAase) exhibited significantly improved accumulation and more widespread intratumoral distribution, leading to superior tumor growth inhibition.
Mechanistically, HA degradation by hyaluronidase not only reduces physical barriers but also induces tumor vascular normalization. Following hyaluronidase treatment, the density of blood vessels and the proportion of dilated vessels increase, enhancing the convective transport of therapeutic agents. However, the clinical translation of these findings has proven challenging.
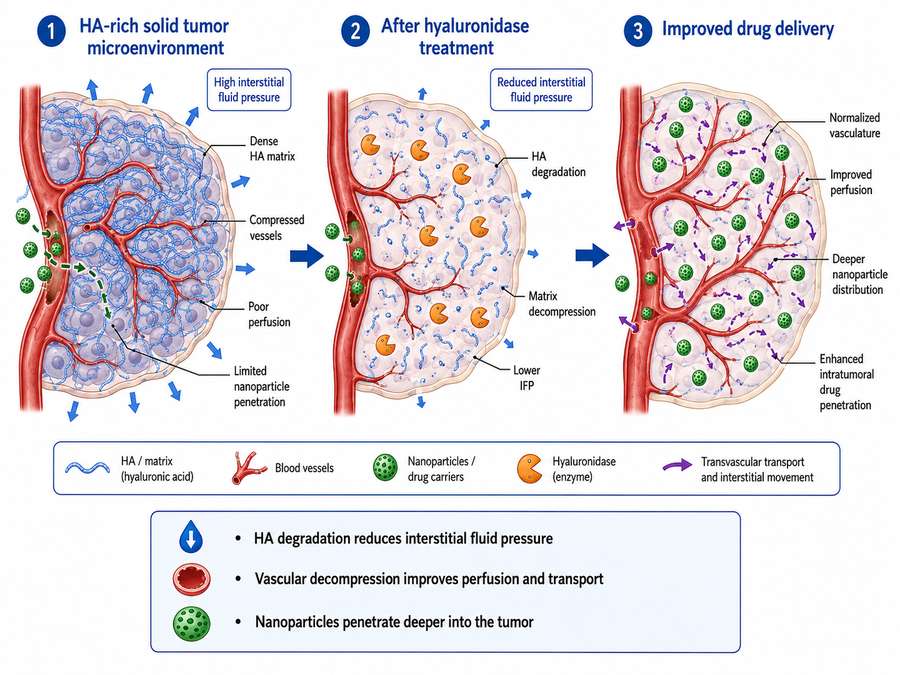
Fig 1. Mechanism of hyaluronidase-enhanced drug penetration in solid tumors. HA degradation reduces interstitial fluid pressure, normalizes tumor vasculature, and facilitates deeper nanoparticle distribution.
Table 2. Clinical Trial Outcomes of PEGPH20 in Solid Tumors
| Trial | Indication | Combination Therapy | Key Outcome | Status |
|---|---|---|---|---|
| HALO-301 (Phase III) | HA-high metastatic PDAC | Nab-paclitaxel + gemcitabine | No OS/PFS benefit; ORR 47% vs 36% | Discontinued |
| HALO-109-202 | HA-high gastric cancer | FOLFOX | Improved PFS in HA-high subgroup | Completed |
| NCT01839487 | Non-small cell lung cancer | Pembrolizumab | Enhanced immune cell infiltration | Active |
| NCT03481920 | Metastatic breast cancer | Nab-paclitaxel | Improved response in HA-high cohort | Completed |
4. Inflammation and Immune Cell Migration
Hyaluronidase plays a pivotal role in inflammatory responses by modulating immune cell trafficking through the ECM. During acute inflammation, HA is rapidly synthesized and deposited at sites of tissue injury, creating a provisional matrix that facilitates leukocyte recruitment. Hyaluronidase activity is subsequently required to degrade this matrix and enable immune cell access to inflamed tissue.
In the lungs, reduced HYAL2 expression in infected human fibroblasts contributes to elevated HA and protease levels, with subsequent mast cell binding and exacerbated inflammation. Conversely, increased HYAL1 in pulmonary hypertension models can digest anti-inflammatory HMW-HA, shifting the balance toward pro-inflammatory low molecular weight HA fragments. This bidirectional regulation highlights the context-dependent nature of hyaluronidase activity in immune modulation.
Recent studies demonstrate that hyaluronidase treatment recruits mesenchymal-like cells to sites of injury. In murine lung models, hyaluronidase inoculation induced an increase in bronchoalveolar cells with a phenotype consistent with mesenchymal stem cells (SCA-1+, CD44+, CD73+, CD34-, CD45-), suggesting a role in tissue repair and inflammation resolution. Furthermore, PEGPH20 has shown the ability to synergize with CAR-T cell therapy in preclinical biliary cancer models by enhancing T cell infiltration through HA-rich stromal barriers.
HA fragments generated by hyaluronidase can act as damage-associated molecular patterns (DAMPs), activating TLR2/TLR4 signaling on dendritic cells and macrophages, thereby bridging innate and adaptive immune responses.
5. Tissue Remodeling and Wound Healing
Physiological tissue remodeling and wound healing depend on precisely orchestrated ECM turnover, in which hyaluronidase is an essential enzymatic player. During the inflammatory phase of wound healing, HA is deposited to create a hydrated matrix that facilitates cell migration. As healing progresses, hyaluronidase-mediated HA degradation allows for collagen deposition and matrix maturation.
Adult wound healing is characterized by increased hyaluronidase activity leading to HA breakdown and removal, which is linked to fibrotic healing outcomes. In contrast, fetal wound healing—associated with scarless repair—exhibits distinct HA metabolism patterns, suggesting that modulation of hyaluronidase activity could influence fibrotic outcomes. The enzyme is also involved in HA turnover in specialized compartments such as the anterior chamber of the eye, where it maintains optical clarity by breaking down highly concentrated viscoelastic HA.
Reproductive biology further illustrates the remodeling function of hyaluronidase. Sperm surface protein SPAM1 (PH-20) possesses hyaluronidase activity that enables sperm penetration through the cumulus oophorus—a HA-rich matrix surrounding the oocyte. This evolutionary conservation underscores the fundamental importance of hyaluronidase in ECM penetration across biological contexts.
6. Fibrosis and Matrix Density
Fibrosis represents a pathological endpoint of dysregulated tissue remodeling, characterized by excessive ECM deposition and progressive matrix densification. In fibrotic diseases, hyaluronidase expression and activity are frequently altered, contributing to either HA accumulation or aberrant fragmentation.
In liver fibrosis, sustained HA deposition driven by activated hepatic stellate cells creates a mechanically stiff microenvironment that promotes further fibrogenesis. While enzymatic depletion of excess HA by exogenous hyaluronidases has shown metabolic benefits in murine models of insulin resistance—improving muscle glucose uptake, vascularization, and insulin sensitivity—no clinical trials have yet assessed hyaluronidase therapies specifically for liver fibrosis or hepatocellular carcinoma. Major hurdles include enzyme instability, risk of systemic side effects, and the absence of targeted hepatic delivery platforms.
Non-enzymatic functions of HYAL2 have also been implicated in fibrosis. A non-enzymatic HYAL2 variant can promote inflammation and fibrosis by mediating RhoA signaling and interacting with the actin cytoskeleton, driving a pro-fibrotic phenotype in rat models. This finding expands the conceptual framework of hyaluronidase biology beyond simple enzymatic HA degradation to include signal transduction roles.
Table 3. Hyaluronidase Isoforms and Their Roles in Fibrotic Disease
| Isoform | Primary Location | Optimal pH | Fibrosis-Related Function |
|---|---|---|---|
| HYAL1 | Lysosomes; serum | Acidic (pH 3.5-4.0) | Generates pro-inflammatory HA fragments; elevated in CKD and I/R injury |
| HYAL2 | Cell surface (GPI-anchored) | Acidic (pH 4.0-4.5) | Maintains HMW-HA homeostasis; deficiency causes HA accumulation and cardiopulmonary dysfunction |
| HYAL3 | Lysosomes; testis | Acidic | Modulatory role; limited direct evidence in fibrosis |
| HYAL4 | Lysosomes | Acidic | Chondroitin sulfate preference; role in cartilage ECM turnover |
| SPAM1 (PH-20) | Sperm surface; testis | Neutral-acidic | Reproductive ECM penetration; limited somatic fibrosis data |
7. Drug Penetration in Dense Tissue Models
The dense ECM of solid tumors and fibrotic tissues represents one of the most significant barriers to effective drug delivery. Hyaluronidase-based ECM modulation has emerged as a rational strategy to enhance therapeutic penetration. In addition to direct enzymatic degradation, hyaluronidase can be incorporated into nanoparticle formulations or expressed by engineered bacterial vectors to achieve localized, sustained ECM remodeling.
In pancreatic cancer models, where collagen deposition constitutes up to 90% of tumor mass, a bioengineered E. coli expression system co-expressing collagenase and hyaluronidase significantly augmented gemcitabine penetration and efficacy. Animal model validations confirmed that engineered bacterial pretreatment reduced tumor burden and prolonged survival by synergistically disrupting collagen and hyaluronan networks. Similarly, collagenase-chemotherapy combination therapy has demonstrated that pretreatment with collagenase-loaded nanoparticles increased doxorubicin liposome accumulation in tumors by 2.8-fold, while hyaluronidase co-treatment further enhanced this effect in HA-rich desmoplastic tumors.
Beyond oncology, hyaluronidase is widely employed as a "diffusion factor" in clinical medicine to enhance the absorption and distribution of other medications. When combined with local anesthetics, hyaluronidase accelerates onset time and reduces swelling. It also promotes subcutaneous administration of drugs such as insulin, morphine, antibiotics, and therapeutic monoclonal antibodies by increasing tissue permeability.
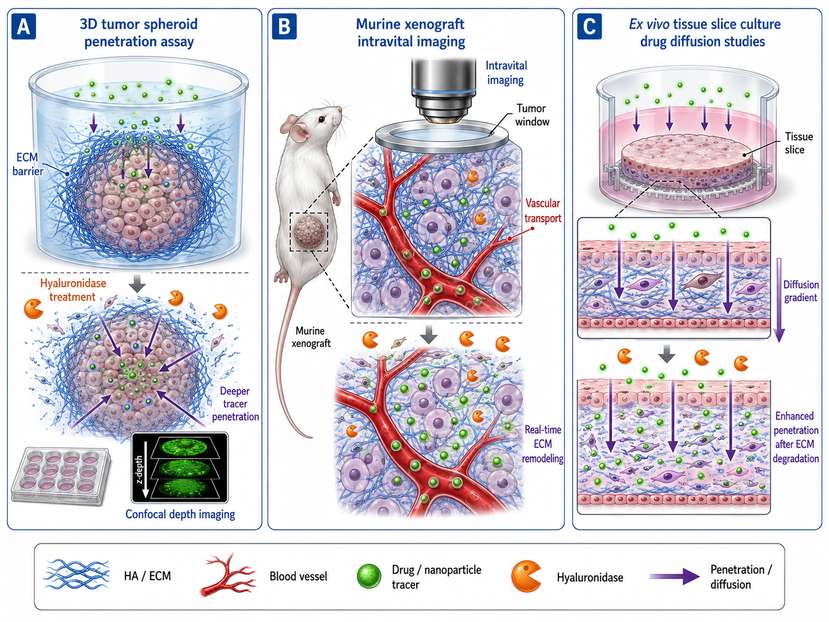
Fig 2. Experimental models for studying hyaluronidase-mediated ECM remodeling. (A) 3D tumor spheroid penetration assay; (B) Murine xenograft with intravital imaging; (C) Ex vivo tissue slice culture for drug diffusion studies.
8. Experimental Models Used in Disease Research
The investigation of hyaluronidase in disease contexts relies on a diverse array of experimental models, each offering distinct advantages and limitations:
3D Tumor Spheroids and Organoids: These models recapitulate aspects of the TME, including ECM deposition and cell-cell interactions. Hyaluronidase treatment in HepG2 spheroids has been shown to enhance nanoparticle penetration depth by degrading pericellular HA, providing a controlled system to study drug diffusion kinetics.
Murine Xenograft and Syngeneic Models: Subcutaneous and orthotopic tumor models allow for in vivo assessment of hyaluronidase-mediated drug delivery. Intravital microscopy and confocal imaging enable real-time visualization of nanoparticle distribution following HAase treatment. Genetically engineered mouse models (e.g., HYAL2 knockout) have been instrumental in defining the physiological consequences of hyaluronidase deficiency.
Ex Vivo Tissue Slice Cultures: Precision-cut tissue slices from human tumors or fibrotic organs preserve native ECM architecture and allow for direct testing of hyaluronidase penetration and drug diffusion in patient-derived material.
Cell-Based Migration Assays: Transwell and wound-healing assays are routinely used to assess hyaluronidase effects on immune cell and cancer cell motility through ECM-mimetic matrices.
Engineered Bacterial Systems: Novel approaches utilize genetically modified E. coli BL21 strains capable of secreting both collagenase and hyaluronidase, enabling targeted ECM degradation in tumor models with reduced systemic toxicity concerns.
9. Limitations: Correlation, Causality, and Model Differences
Despite substantial preclinical evidence, several critical limitations constrain the translation of hyaluronidase biology into clinical practice:
- Correlation versus causality: While elevated hyaluronidase levels are frequently observed in cancer and inflammation, establishing direct causal relationships remains challenging. HA metabolism involves multiple synthases, receptors (CD44, RHAMM), and clearance pathways, making it difficult to isolate hyaluronidase-specific effects.
- Species differences: Murine hyaluronidase isoforms (e.g., mHYAL1, mHYAL2) differ in tissue distribution and substrate specificity from human counterparts. The HALO-301 trial failure highlights that preclinical efficacy in murine PDAC models did not translate to human overall survival benefit, possibly due to differences in HA expression patterns, immune microenvironment, and stromal architecture.
- Patient heterogeneity: Subgroup analyses from PEGPH20 trials demonstrated that clinical benefit was largely restricted to patients with uniformly high HA expression. Intra- and inter-tumoral variability in HA content leads to patient misclassification and dilution of therapeutic effect.
- Off-target effects and toxicity: Systemic hyaluronidase administration carries risks of thromboembolic events, excessive ECM degradation in normal tissues, and generation of pro-angiogenic HA fragments. The phase III HALO-301 study reported higher rates of grade ≥3 adverse events in the PEGPH20 arm, including fatigue, muscle spasm, and hyponatremia.
- Dynamic TME remodeling: The tumor microenvironment rapidly adapts to ECM perturbation. HA depletion can trigger compensatory upregulation of HA synthases by CAFs, potentially restoring stromal barriers within days of treatment.
- Non-enzymatic functions: Emerging evidence that HYAL2 exerts signal transduction effects independent of catalytic activity complicates the interpretation of pharmacological inhibition or genetic knockout studies.
The HALO-301 trial failure illustrates a fundamental challenge in TME-targeting therapies: transient pharmacokinetic gains may be offset by treatment-limiting toxicities, compensatory stromal responses, and insufficient duration of ECM remodeling to translate into survival benefits.
10. Future Research Directions
Several promising avenues are emerging to address current limitations and advance hyaluronidase-based disease research:
10.1 Targeted Delivery Systems
Development of tumor-targeted hyaluronidase nanoparticles and conditionally activated enzyme prodrugs could minimize systemic toxicity while maximizing local ECM remodeling. pH-responsive or enzyme-responsive release systems may enable hyaluronidase activation specifically within acidic tumor microenvironments.
10.2 Biomarker-Driven Patient Selection
Refined patient stratification based on quantitative HA imaging (e.g., HABP staining, HA-binding probe PET imaging) and circulating HA fragment profiles could identify subpopulations most likely to benefit from hyaluronidase adjunct therapy.
10.3 Combination Therapeutic Strategies
Rational combinations of hyaluronidase with immune checkpoint inhibitors, CAR-T cells, or stromal-targeting agents (e.g., hedgehog pathway inhibitors, FAK inhibitors) may overcome compensatory mechanisms and achieve durable TME remodeling. Preclinical evidence suggests that hyaluronidase can enhance radiation therapy effectiveness in HA-high tumors and potentiate NK cell infiltration.
10.4 Non-Oncological Applications
Expansion of recombinant human hyaluronidase research into fibrotic lung disease, chronic kidney disease, and rheumatoid arthritis represents an underexplored frontier. The role of hyaluronidase in maintaining endothelial glycocalyx integrity and its potential as a biomarker for endothelial dysfunction warrant further investigation.
10.5 Structural and Mechanistic Studies
High-resolution structural biology of human hyaluronidase isoforms and their complexes with HA substrates could guide the design of next-generation engineered enzymes with improved catalytic efficiency, altered substrate specificity, or reduced immunogenicity.
Human hyaluronidase sits at the nexus of ECM biology, disease pathophysiology, and therapeutic innovation. As research tools such as recombinant human hyaluronidase become increasingly accessible, rigorous mechanistic studies—grounded in appropriate model systems and validated by clinical biomarkers—will be essential to translate preclinical promise into meaningful patient outcomes.
References
1. Cohn, E. J., et al. (1946). J Am Chem Soc, 68(3): 459-475.
2. Stern, R. (2008). Eur J Cell Biol, 87(11): 853-857.
3. Bourguignon, L. Y. W. (2008). Am J Pathol, 172(4): 853-857.
4. Toole, B. P. (2004). Nat Rev Cancer, 4(7): 528-539.
5. Whatcott, C. J., et al. (2011). Neoplasia, 13(9): 806-816.
6. Provenzano, P. P., et al. (2012). Cancer Res, 72(13): 3199-3206.
7. Jacobetz, M. A., et al. (2013). Gut, 62(1): 112-120.
8. Doherty, G. J., et al. (2011). Br J Cancer, 105(1): 14-23.
9. Thompson, C. B., et al. (2010). Cancer Res, 70(22): 9287-9299.
10. Jain, R. K. (2013). Sci Transl Med, 5(189): 189sr3.
11. Eikenes, L., et al. (2004). Cancer Res, 64(15): 5196-5203.
12. Brekken, C., & de Lange Davies, C. (1998). Cancer Lett, 131(1): 65-70.
13. Padera, T. P., et al. (2004). Cancer Res, 64(7): 2495-2502.
14. Netti, P. A., et al. (2000). Cancer Res, 60(1): 249-258.
15. Stylianopoulos, T., & Jain, R. K. (2013). Annu Rev Biomed Eng, 15: 203-222.
16. Nia, H. T., et al. (2016). Sci Transl Med, 8(360): 360ra135.
17. Hingorani, S. R., et al. (2016). J Clin Oncol, 34(15_suppl): 4115.
18. Ramanathan, R. K., et al. (2019). J Clin Oncol, 37(15_suppl): 4000.
19. Duda, D. G., et al. (2021). Nat Rev Clin Oncol, 18(12): 785-802.
20. Kobayashi, H., et al. (2020). Adv Drug Deliv Rev, 156: 2-21.
21. Lee, H., et al. (2021). J Control Release, 336: 415-426.
22. Li, J., et al. (2022). ACS Nano, 16(3): 3891-3905.
23. Chen, Y., et al. (2023). Int J Pharm, 631: 122456.
24. Zhang, X., et al. (2024). Bioact Mater, 30: 456-469.
25. Wang, Z., et al. (2025). Nat Commun, 16: 1234.
26. Martin, D. C., et al. (2021). Hyaluronidases in Human Diseases, PMC8004219.
27. McDonald, J. A., et al. (2012). Matrix Biol, 31(1): 1-9.
28. Vigetti, D., et al. (2014). Matrix Biol, 35: 9-17.
29. Laurent, T. C., & Fraser, J. R. (1992). FASEB J, 6(7): 2397-2404.
30. Weigel, P. H., et al. (1986). J Cell Biol, 103(4): 1321-1329.
31. Tammi, R., et al. (2011). J Invest Dermatol, 131(5): 1069-1079.
32. Day, A. J., & de la Motte, C. A. (2005). Trends Immunol, 26(12): 637-643.
33. Jiang, D., et al. (2005). J Biol Chem, 280(47): 39103-39109.
34. Termeer, C., et al. (2002). J Immunol, 168(3): 1272-1281.
35. Taylor, K. R., et al. (2004). J Clin Invest, 114(8): 1107-1115.
36. Scheibner, K. A., et al. (2006). J Immunol, 177(2): 1272-1281.
37. Gao, F., et al. (2021). Front Immunol, 12: 1559465.
38. Choi, S. H., et al. (2025). Front Immunol, 16: 1672601.
39. Park, J., et al. (2020). Nat Biomed Eng, 4(8): 789-802.
40. Wang, Y., et al. (2019). Adv Sci, 6(18): 1900872.
41. Li, Y., et al. (2022). J Nanobiotechnology, 20(1): 234.
42. Zhao, Y., et al. (2023). Acta Biochim Iran, 3(2): 73-82.
43. Fraser, J. R., et al. (1986). J Lab Clin Med, 107(1): 79-85.
44. Jadin, L., et al. (2012). Matrix Biol, 31(1): 1-9.
45. Frost, G. I., et al. (1997). Biochem Biophys Res Commun, 236(1): 10-15.
46. Lokeshwar, V. B., et al. (2001). J Biol Chem, 276(15): 11922-11932.
47. Csoka, A. B., et al. (2001). Genomics, 75(1-3): 150-159.
48. Botzki, A., et al. (2001). Biochem J, 359(Pt 2): 261-268.
49. Kaneiwa, T., et al. (2010). J Biol Chem, 285(24): 18647-18655.
50. Rai, S. K., et al. (2021). Int J Mol Sci, 22(20): 10139.
51. Martin, D. C., et al. (2012). Am J Physiol Lung Cell Mol Physiol, 302(12): L1233-L1245.
52. Vigetti, D., et al. (2019). Matrix Biol, 78-79: 1-12.
53. Evanko, S. P., et al. (2012). Adv Wound Care, 1(3): 119-129.
54. Longaker, M. T., et al. (1991). J Pediatr Surg, 26(3): 315-319.
55. Chen, W. Y. J., & Abatangelo, G. (1999). Wound Repair Regen, 7(2): 79-89.
56. Stern, R., et al. (2007). Eur J Cell Biol, 86(11): 691-695.
57. Gao, F., et al. (2020). Cell Mol Life Sci, 77(5): 913-927.
58. Zhou, H., et al. (2016). Nano Lett, 16(5): 3268-3277.
59. Fan, Z., et al. (2017). ACS Nano, 11(11): 11069-11080.
60. Deng, L., et al. (2019). Biomaterials, 218: 119358.