The Glucocerebrosidase Pathway and Its Role in Lipid Metabolism
Understanding Sphingolipid Metabolism and GBA Enzyme Function in Cellular Homeostasis
Abstract
The glucocerebrosidase (GBA) pathway represents a critical node in sphingolipid metabolism, catalyzing the hydrolysis of glucocerebroside to glucose and ceramide within lysosomal compartments. This enzymatic reaction maintains cellular lipid homeostasis and prevents the pathological accumulation of glycosphingolipids. Mutations in the GBA gene disrupt this pathway, leading to Gaucher disease and significantly increasing the risk for Parkinson's disease. This pathway analysis examines the biochemical mechanisms of GBA-mediated lipid processing, the consequences of pathway disruptions, and emerging therapeutic strategies targeting this critical metabolic axis. Understanding the imiglucerase pathway provides essential insights for both basic research and clinical applications.
glucocerebrosidase pathway, sphingolipid metabolism, GBA enzyme, lysosomal storage disease, lipid homeostasis, Gaucher disease
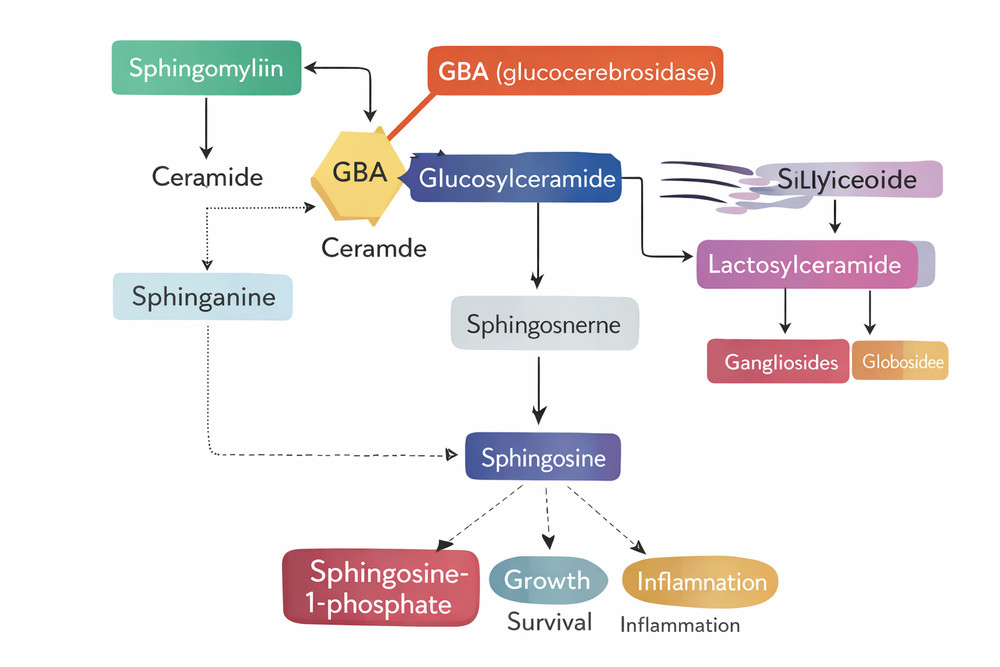
Fig 1. Overview of sphingolipid metabolic pathways highlighting the GBA enzyme position
1. Overview of Sphingolipid Metabolism
Sphingolipids constitute a diverse class of lipids essential for membrane structure, cell signaling, and protein trafficking. The metabolic network begins with the condensation of serine and palmitoyl-CoA in the endoplasmic reticulum, forming 3-ketosphinganine, which is subsequently reduced to dihydrosphingosine. Through N-acylation and desaturation, ceramide emerges as the central hub of sphingolipid metabolism.
Ceramide serves as the precursor for complex sphingolipids including sphingomyelin, glycosphingolipids, and gangliosides. These molecules are transported to the Golgi apparatus and plasma membrane via vesicular trafficking. The catabolic arm of this network occurs primarily within lysosomes, where specific hydrolases sequentially degrade complex sphingolipids back to ceramide and ultimately to sphingosine and fatty acids.
1.1 The Lysosomal Sphingolipid Degradation Cascade
Lysosomal degradation of glycosphingolipids follows a strict hierarchical order. Glycosidases remove terminal sugar residues sequentially until ceramide remains. Glucocerebrosidase (acid β-glucosidase, GBA) occupies a critical position in this cascade, catalyzing the penultimate step before ceramide release. The enzyme hydrolyzes the β-glycosidic bond between glucose and ceramide, generating ceramide that is subsequently cleaved by acid ceramidase.
1.2 Interconnection with Other Metabolic Pathways
Sphingolipid metabolism intersects with multiple cellular processes. The ceramide generated by GBA activity can be phosphorylated to ceramide-1-phosphate, a signaling molecule regulating cell survival and inflammation. Alternatively, ceramide conversion to sphingosine and subsequent phosphorylation produces sphingosine-1-phosphate (S1P), a potent bioactive lipid mediating cell migration, angiogenesis, and immune responses.
| Metabolic Step | Enzyme | Substrate | Product | Cellular Location |
|---|---|---|---|---|
| Ganglioside catabolism | β-hexosaminidase A | GM2 ganglioside | GM3 ganglioside | Lysosome |
| Lactosylceramide cleavage | β-galactosidase | Lactosylceramide | Glucosylceramide | Lysosome |
| Glucosylceramide hydrolysis | Glucocerebrosidase (GBA) | Glucosylceramide | Ceramide + Glucose | Lysosome |
| Ceramide degradation | Acid ceramidase | Ceramide | Sphingosine + Fatty acid | Lysosome |
| Sphingosine recycling | Sphingosine kinase | Sphingosine | Sphingosine-1-phosphate | Cytosol |
2. Role of GBA Enzyme in Lipid Homeostasis
2.1 Structural and Biochemical Properties
Glucocerebrosidase (GBA, EC 3.2.1.45) is a 497-amino acid glycoprotein synthesized as a precursor in the endoplasmic reticulum. The mature enzyme contains three N-linked glycosylation sites and requires proper folding and trafficking to lysosomes via the mannose-6-phosphate receptor pathway. The enzyme's catalytic mechanism involves acid-base catalysis with two critical glutamate residues (E235 and E340) positioned in the active site.
GBA requires saposin C as an essential cofactor for optimal activity. This small glycoprotein, derived from prosaposin proteolytic processing, binds to both the enzyme and its lipid substrate, presenting glucosylceramide in a conformation accessible to the catalytic machinery. The pH optimum of GBA activity is 5.5, consistent with its lysosomal localization.
2.2 Catalytic Mechanism and Kinetic Parameters
The enzymatic reaction follows a retaining mechanism where the anomeric configuration of the glucose moiety is preserved. The catalytic efficiency (kcat/Km) of wild-type GBA for natural substrate glucosylceramide is approximately 10^5 M^-1 s^-1. The enzyme demonstrates broad specificity for acyl chain lengths but shows highest affinity for C16:0 and C18:0 fatty acid-containing substrates, reflecting the composition of cellular membranes.
GBA exhibits maximum catalytic efficiency at pH 5.5 with saposin C as essential cofactor. The enzyme processes approximately 100-200 substrate molecules per minute under saturating conditions.
2.3 Cellular Functions Beyond Lipid Degradation
Emerging evidence indicates GBA participates in multiple cellular processes beyond its canonical lysosomal function. The enzyme interacts with α-synuclein in lipid rafts, potentially influencing protein aggregation pathways. Reduced GBA activity correlates with lysosomal dysfunction, impaired autophagy, and mitochondrial abnormalities. These pleiotropic effects explain why GBA mutations confer risk for both Gaucher disease and Parkinson's disease.
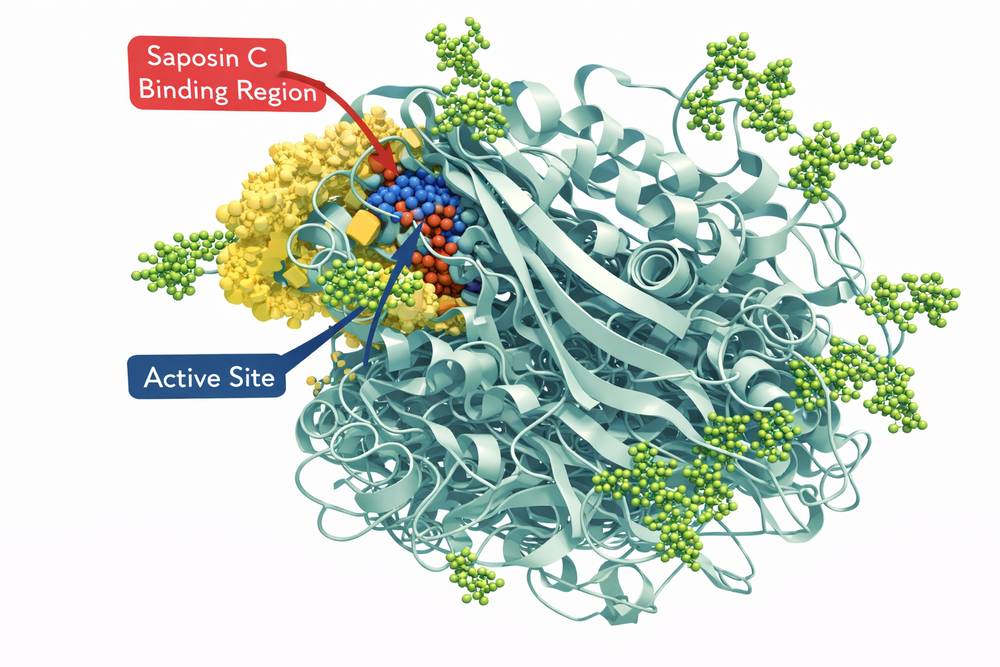
Fig 2. Three-dimensional structure of glucocerebrosidase showing active site and saposin C binding region
3. Pathway Disruptions and Genetic Variants
3.1 Gaucher Disease: The Prototypical GBA Deficiency
Gaucher disease represents the most common lysosomal storage disorder, resulting from biallelic GBA mutations causing enzyme deficiency. The disease manifests in three clinical subtypes based on neurological involvement. Type 1 (non-neuronopathic) accounts for 90% of cases and presents with hepatosplenomegaly, cytopenias, and bone complications. Types 2 and 3 involve central nervous system pathology with variable severity.
Over 400 GBA mutations have been identified, ranging from severe null alleles to milder missense variants. The N370S mutation is most prevalent in Ashkenazi Jewish populations, while L444P occurs worldwide and associates with neuronopathic forms. Genotype-phenotype correlations remain imperfect due to modifying genetic and environmental factors.
3.2 GBA-Associated Parkinson's Disease
Heterozygous GBA mutations constitute the most significant genetic risk factor for Parkinson's disease, increasing susceptibility 5-20-fold depending on the specific variant. The mechanism involves loss of enzyme function leading to glucosylceramide accumulation, lysosomal stress, and α-synuclein aggregation. This connection establishes the GBA pathway as a therapeutic target for synucleinopathies.
| GBA Variant | Enzyme Activity (%) | Associated Phenotype | Population Frequency | Therapeutic Considerations |
|---|---|---|---|---|
| N370S (c.1226A>G) | 45-60% | Type 1 Gaucher; Parkinson's risk | 1/850 (Ashkenazi) | ERT responsive |
| L444P (c.1448T>C) | 10-20% | Neuronopathic Gaucher; High PD risk | 1/2000 (Global) | Substrate reduction therapy |
| 84GG (c.84dupG) | 0% (null allele) | Severe Type 2 Gaucher | Rare | HSCT or gene therapy |
| E326K (c.1093G>A) | 70-80% | Moderate PD risk | 1/100 (Various) | Monitoring recommended |
| T369M (c.1223C>T) | 85-95% | Asymptomatic carrier | 1/50 (Ashkenazi) | Genetic counseling |
3.3 Secondary Pathway Impairments
GBA dysfunction creates a cascade of metabolic disturbances. Accumulated glucosylceramide and glucosylsphingosine alter macrophage activation, promoting inflammatory cytokine release. The lipid storage in tissue macrophages produces characteristic "Gaucher cells" with distinctive crumpled tissue paper cytoplasm. Bone marrow infiltration leads to osteopenia, avascular necrosis, and pathological fractures.
4. Cellular Consequences of GBA Pathway Dysfunction
4.1 Lysosomal Dysfunction and Autophagy Impairment
GBA deficiency compromises lysosomal integrity through multiple mechanisms. Accumulated lipids alter lysosomal membrane composition, reducing membrane fluidity and impairing fusion with autophagosomes. The resulting autophagic block leads to accumulation of damaged mitochondria and protein aggregates. Studies demonstrate that even partial GBA reduction (as in heterozygous carriers) produces measurable lysosomal abnormalities.
4.2 Mitochondrial Dysfunction and Oxidative Stress
Impaired autophagy prevents clearance of damaged mitochondria, leading to accumulation of defective organelles with compromised membrane potential. These mitochondria generate excessive reactive oxygen species (ROS), further damaging cellular components. The resulting oxidative stress exacerbates protein misfolding and lipid peroxidation, creating a self-amplifying cycle of cellular damage.
4.3 Inflammation and Immune Dysregulation
Glucosylceramide-engorged macrophages adopt a pro-inflammatory phenotype, secreting IL-1β, IL-6, and TNF-α through inflammasome activation. Chronic inflammation contributes to tissue damage in Gaucher disease and may accelerate neurodegeneration in Parkinson's disease. The immune dysregulation also explains the increased risk of hematologic malignancies and multiple myeloma observed in Gaucher patients.
4.4 Neuronal Vulnerability
Neurons are particularly susceptible to GBA dysfunction due to their high metabolic demands and complex lysosomal trafficking requirements. Dopaminergic neurons of the substantia nigra show selective vulnerability, likely related to their extensive axonal arbors and high oxidative metabolism. GBA deficiency promotes α-synuclein aggregation through both direct lipid-mediated mechanisms and indirect lysosomal stress pathways.
5. Therapeutic Targeting of the GBA Pathway
5.1 Enzyme Replacement Therapy (ERT)
Intravenous administration of recombinant glucocerebrosidase revolutionized Type 1 Gaucher disease treatment. Early preparations used placental tissue, but current products utilize Chinese hamster ovary (CHO) cells expressing human GBA with optimized glycosylation patterns for macrophage targeting. Imiglucerase products demonstrate remarkable efficacy in reversing visceral manifestations, with 90% reduction in liver and spleen volume within 12-24 months.
However, ERT limitations include inability to cross the blood-brain barrier, necessitating alternative approaches for neuronopathic forms. Infusion-related reactions and antibody development occur in 10-15% of patients, requiring premedication or dose adjustments.
5.2 Substrate Reduction Therapy (SRT)
Small molecule inhibitors of glucosylceramide synthase (the enzyme producing GBA substrate) offer an oral alternative to ERT. Miglustat and eliglustat reduce substrate burden by limiting de novo synthesis. SRT is particularly valuable for patients with antibodies to ERT or those preferring oral therapy. Eliglustat, a potent specific inhibitor, shows non-inferiority to ERT for Type 1 Gaucher disease.
5.3 Pharmacological Chaperones
Mutant GBA proteins often misfold and are retained in the endoplasmic reticulum, preventing lysosomal delivery. Pharmacological chaperones bind to the enzyme active site, stabilizing proper conformation and promoting trafficking. Ambroxol, an established expectorant with chaperone activity, increases GBA activity in patient cells and has entered clinical trials for Parkinson's disease. Next-generation chaperones with improved specificity are in development.
5.4 Gene Therapy Approaches
Lentiviral and adeno-associated viral (AAV) vectors delivering functional GBA genes show promise in preclinical models. Direct CNS delivery via intrathecal injection addresses the neurological limitations of ERT. Hematopoietic stem cell gene therapy offers the theoretical advantage of permanent correction with single treatment. Clinical trials are evaluating safety and efficacy of these advanced modalities.
| Therapeutic Approach | Mechanism | Indications | Route | Key Advantages |
|---|---|---|---|---|
| Enzyme Replacement (ERT) | Exogenous GBA supplementation | Type 1 Gaucher | Intravenous | Established efficacy; reversible |
| Substrate Reduction (SRT) | GCS inhibition | Type 1 Gaucher; Adjunct therapy | Oral | Convenience; CNS penetration |
| Pharmacological Chaperones | Protein folding correction | Amenable mutants; Parkinson's | Oral | Addresses ER retention; low cost |
| Gene Therapy | Permanent GBA expression | All types; Severe cases | IV/Intrathecal | Single treatment; CNS delivery |
| HSCT | Donor macrophage replacement | Neuronopathic types | Transplantation | Permanent correction; CNS access |
For Type 1 Gaucher disease, enzyme replacement therapy with imiglucerase remains the first-line treatment with proven long-term efficacy. Substrate reduction therapy offers a valuable alternative for appropriate candidates. Emerging chaperone and gene therapies promise to address current limitations in CNS delivery.
6. Conclusion
The glucocerebrosidase pathway serves as a nexus connecting lipid metabolism, lysosomal function, and neurodegenerative disease. Understanding the intricate regulation of this pathway has transformed our approach to Gaucher disease and opened new therapeutic avenues for Parkinson's disease. The convergence of basic biochemistry, cell biology, and clinical medicine exemplified by GBA research provides a model for translational science.
Future directions include development of brain-penetrant enzyme formulations, precision medicine approaches based on specific mutations, and combination therapies targeting multiple pathway nodes. As our understanding of sphingolipid signaling expands, the GBA pathway will likely reveal additional connections to human disease, further emphasizing the importance of this metabolic axis in health and pathology.
References
1. Grabowski, G. A., et al. (2020). Lancet, 396(10248): 1315-1326.
2. Sidransky, E., et al. (2009). N Engl J Med, 361(17): 1651-1661.
3. Schapira, A. H. V. (2015). J Parkinsons Dis, 5(3): 449-458.
4. Mistry, P. K., et al. (2017). Am J Hematol, 92(2): 190-198.
5. Hughes, D., et al. (2022). Mol Genet Metab, 135(2): 118-125.