Gaucher Disease: Molecular Basis and Research Models
Comprehensive Disease Background Resource for Researchers and Clinicians
Abstract
Gaucher disease (GD) represents the most prevalent lysosomal storage disorder, arising from autosomal recessive mutations in the GBA1 gene that encodes glucocerebrosidase (GCase). This comprehensive resource examines the molecular foundations of GD, including genetic mechanisms, biochemical pathways, disease phenotypes, and research models. We present detailed analysis of over 400 identified GBA1 mutations, with particular focus on prevalent variants such as N370S and L444P. The document explores three clinical subtypes—non-neuronopathic (Type 1), acute neuronopathic (Type 2), and subacute neuronopathic (Type 3)—and their underlying pathophysiological mechanisms. Furthermore, we review current and emerging therapeutic strategies, including enzyme replacement therapy (ERT), substrate reduction therapy (SRT), pharmacological chaperone therapy (PCT), and gene therapy approaches. This resource serves as a foundational reference for researchers investigating Gaucher disease mechanism and GBA mutation research.
Gaucher disease mechanism, GBA mutation research, glucocerebrosidase, lysosomal storage disorder, enzyme replacement therapy, imiglucerase products
1. Genetic Background of Gaucher Disease
1.1 GBA1 Gene and Inheritance Pattern
Gaucher disease is inherited in an autosomal recessive manner, with causative mutations located in the GBA1 gene on chromosome 1q21. This gene spans approximately 7.5 kb and comprises 11 exons encoding the 497-amino acid glucocerebrosidase enzyme. The GBA1 gene is accompanied by a highly homologous pseudogene, GBAP1, located 16 kb downstream, which complicates genetic analysis due to potential gene conversion events and recombination.
To date, researchers have identified over 400 distinct pathogenic variants in the GBA1 gene, including missense mutations, nonsense mutations, frameshifts, splice site alterations, and complex rearrangements. These mutations result in deficient glucocerebrosidase activity, leading to accumulation of glucosylceramide (GlcCer) and glucosylsphingosine (GlcSph) within lysosomes of macrophages and other cell types.
1.2 Common Pathogenic Variants
Among the extensive catalog of GBA1 mutations, several variants demonstrate higher prevalence and have been extensively characterized in GBA mutation research:
| Mutation | Nucleotide Change | GD Type Association | Residual Activity | Clinical Severity |
|---|---|---|---|---|
| N370S (p.Asn370Ser) | c.1226A>G | Type 1 (Non-neuronopathic) | ~15-20% | Mild to Moderate |
| L444P (p.Leu444Pro) | c.1448T>C | Type 2/3 (Neuronopathic) | ~5-10% | Severe |
| D409H (p.Asp409His) | c.1342G>C | Type 3 (Subacute neuronopathic) | ~10-15% | Moderate to Severe |
| V394L (p.Val394Leu) | c.1180G>C | Type 1/3 | ~10-15% | Variable |
| 84GG (c.84dupG) | Insertion | Type 2 (Acute neuronopathic) | 0% | Very Severe |
The N370S mutation represents the most prevalent GBA1 variant globally, particularly among Ashkenazi Jewish populations where it accounts for approximately 70% of mutant alleles. This mutation is exclusively associated with Type 1 GD and is considered protective against primary neurological involvement. However, recent longitudinal studies reveal that even Type 1 patients carrying N370S mutations exhibit a 5-7% risk of developing Parkinson's disease before age 70, highlighting the complex relationship between GBA1 variants and neurodegenerative processes.
The L444P mutation demonstrates strong association with neuronopathic forms of GD (Types 2 and 3). This variant originated from a founder effect in northern Sweden (Norrbottnian variant) but is now identified globally. Homozygosity for L444P typically results in severe disease with prominent neurological manifestations. Notably, heterozygous carriers of L444P have a significantly increased risk (OR = 20.2) of developing Parkinson's disease compared to non-carriers, underscoring the importance of Gaucher disease mechanism in understanding synucleinopathies.
1.3 Genotype-Phenotype Correlations
Understanding the relationship between genetic variants and clinical presentation remains complex. While certain patterns exist—such as N370S with Type 1 and L444P with neuronopathic forms—clinical expression is influenced by multiple factors:
- Allelic heterogeneity: Compound heterozygotes often display intermediate phenotypes between the two mutations
- Modifier genes: Variants in GBA2, prosaposin, and other lysosomal genes can modulate disease severity
- Epigenetic factors: DNA methylation patterns and histone modifications affect gene expression
- Environmental influences: Inflammation and metabolic stress can exacerbate symptom presentation
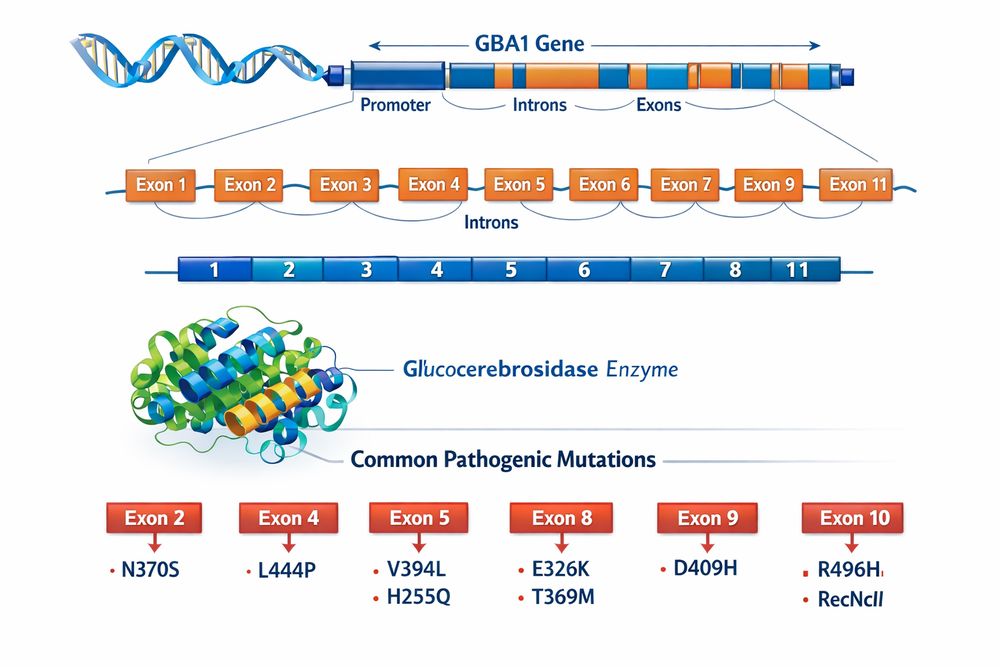
Fig 1. GBA1 gene structure, exon organization, and distribution of common pathogenic mutations
2. Types of Gaucher Disease
Gaucher disease is traditionally classified into three main types based on the presence and severity of neurological involvement. However, contemporary clinical understanding recognizes a phenotypic continuum rather than discrete categories, with considerable overlap between classifications.
| Feature | Type 1 (Non-neuronopathic) | Type 2 (Acute Neuronopathic) | Type 3 (Subacute Neuronopathic) |
|---|---|---|---|
| Neurological Involvement | Absent (traditionally); secondary complications possible | Severe, rapid progression | Present, slowly progressive |
| Age of Onset | Any age (childhood to adulthood) | Infancy (birth to 6 months) | Childhood (before age 20) |
| Life Expectancy | Normal with treatment | <2 years (fatal) | Variable, up to 5th-6th decade |
| Visceral Symptoms | Prominent | Present but overshadowed by CNS | Prominent |
| Common Mutations | N370S/N370S, N370S/Other | 84GG/84GG, L444P/L444P | L444P/L444P, D409H/D409H |
| ERT Response | Excellent | Limited (CNS not accessible) | Partial (visceral only) |
2.1 Type 1 Gaucher Disease (Non-neuronopathic)
Type 1 GD is the most common form, accounting for approximately 95% of cases in Western populations. It is characterized by the absence of primary central nervous system involvement, though patients may experience secondary neurological complications from spinal cord compression due to bone disease. The clinical spectrum ranges from asymptomatic individuals diagnosed incidentally to severely affected patients with massive hepatosplenomegaly, cytopenias, and skeletal complications.
- Splenomegaly (85%): Often massive, causing early satiety and abdominal discomfort
- Thrombocytopenia (68%): Leading to bleeding tendencies and easy bruising
- Hepatomegaly (63%): Usually moderate, rarely causing liver dysfunction
- Bone disease (55%): Including osteopenia, bone pain, crises, and fractures
- Anemia (34%): Resulting from bone marrow infiltration and hypersplenism
2.2 Type 2 Gaucher Disease (Acute Neuronopathic)
Type 2 GD is the rarest and most severe form, occurring in approximately 1% of cases. It presents in infancy with rapid neurological deterioration, typically fatal within the first two years of life. The disease is characterized by early-onset brainstem dysfunction, including:
- Stridor and laryngospasm
- Retroflexion of the head (opisthotonus)
- Swallowing difficulties and aspiration
- Seizures and hypertonia
- Developmental regression
Currently, no disease-modifying therapy exists for Type 2 GD, as enzyme replacement therapy cannot cross the blood-brain barrier effectively.
2.3 Type 3 Gaucher Disease (Subacute Neuronopathic)
Type 3 GD represents an intermediate phenotype affecting approximately 5% of patients. It combines visceral manifestations similar to Type 1 with slowly progressive neurological deterioration. The Norrbottnian variant, prevalent in northern Sweden, is a well-characterized subtype featuring:
- Early massive visceral involvement
- Horizontal supranuclear gaze palsy (pathognomonic)
- Progressive kyphoscoliosis
- Cognitive decline and dementia in later stages
- Myoclonic epilepsy
- Ataxia and spasticity
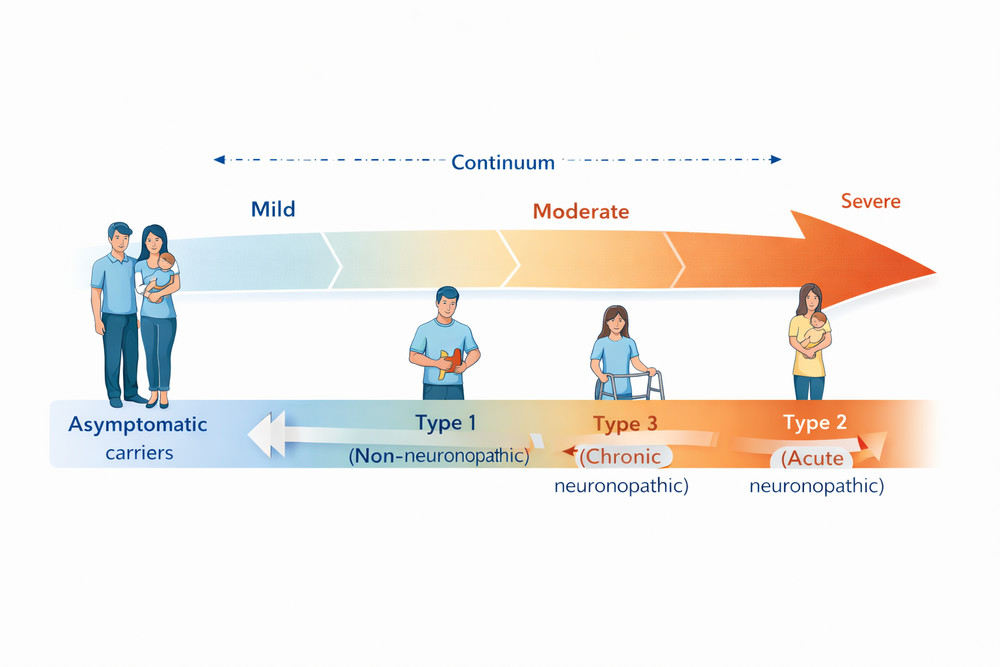
Fig 2. The phenotypic spectrum of Gaucher disease showing the continuum from asymptomatic carriers to severe neuronopathic forms
3. Biochemical Mechanisms
3.1 Glucocerebrosidase Enzyme Function
Glucocerebrosidase (GCase, EC 3.2.1.45) is a lysosomal acid β-glucosidase that catalyzes the hydrolysis of glucosylceramide (GlcCer) into glucose and ceramide. This reaction represents the final step in the degradation pathway of complex glycosphingolipids derived from the turnover of cellular membranes, particularly those of hematopoietic cells.
Glucosylceramide
(GlcCer)
Glucocerebrosidase
(GCase)
Saposin C
(SapC)
Glucose + Ceramide
3.2 Pathophysiological Cascade
The fundamental Gaucher disease mechanism involves accumulation of undegraded substrates within lysosomes of macrophages, transforming them into characteristic "Gaucher cells." These lipid-laden macrophages trigger a complex pathophysiological cascade:
| Pathological Process | Molecular Mechanism | Clinical Consequence |
|---|---|---|
| Substrate Accumulation | GlcCer and GlcSph storage in lysosomes | Gaucher cell formation, organomegaly |
| Lysosomal Dysfunction | Impaired autophagy, calcium dysregulation | Cellular stress, apoptosis |
| Inflammation | IL-1β, IL-6, TNF-α activation; C5a-C5aR1 pathway | Chronic inflammation, bone disease |
| Neurodegeneration | α-synuclein aggregation, ER stress, mitochondrial dysfunction | Parkinsonism, cognitive decline |
| Immune Dysregulation | B-cell hyperproliferation, monoclonal gammopathy | Multiple myeloma risk, infections |
3.3 Toxic Sphingolipids: GlcSph Hypothesis
Recent research has highlighted the importance of glucosylsphingosine (GlcSph, also called lyso-Gb1) as a key toxic metabolite. Unlike GlcCer, GlcSph is not normally present in significant amounts in healthy tissues but accumulates dramatically in GD patients. Elevated GlcSph levels have been associated with:
- Direct cytotoxicity to neurons and bone cells
- Activation of inflammatory pathways
- Impairment of osteoblast function leading to osteoporosis
- Promotion of α-synuclein aggregation in Parkinson's disease
GlcSph has emerged as a promising biomarker for disease severity and therapeutic monitoring, with levels correlating better with clinical outcomes than traditional markers like chitotriosidase.
3.4 GBA1-Parkinson's Disease Connection
The discovery that heterozygous GBA1 mutations are the most common genetic risk factor for Parkinson's disease (PD) has revolutionized our understanding of both conditions. The mechanistic link involves:
1. Lysosomal dysfunction: Reduced GCase activity impairs autophagy-lysosome pathway
2. α-synuclein accumulation: Bidirectional relationship where GCase deficiency promotes α-synuclein aggregation, and aggregated α-synuclein further inhibits GCase
3. ER stress and UPR activation: Mutant GCase misfolding triggers unfolded protein response
4. Mitochondrial dysfunction: Altered sphingolipid metabolism affects mitochondrial dynamics
4. Cell and Animal Models
4.1 In Vitro Models
4.1.1 Patient-Derived Cells
Primary cells from GD patients have been instrumental in understanding disease mechanisms:
- Skin fibroblasts: Classic model for enzyme activity assays and substrate accumulation studies
- Monocyte-derived macrophages: Recapitulate Gaucher cell pathology
- Bone marrow stromal cells: Model for skeletal manifestations
4.1.2 Induced Pluripotent Stem Cells (iPSCs)
Patient-derived iPSCs represent a breakthrough in GD research, enabling differentiation into relevant cell types including:
- Macrophages and microglia for studying storage pathology
- Neurons for modeling Parkinson's disease mechanisms
- Osteoblasts for investigating bone disease
- Hepatocytes for liver involvement studies
CRISPR/Cas9 technology allows the generation of isogenic controls and the introduction of specific mutations to study genotype-phenotype correlations in iPSC models.
4.2 Mouse Models
4.2.1 Knockout (KO) Models
Complete Gba1 knockout in mice results in neonatal lethality within 48 hours due to severe skin barrier defects (collodion baby phenotype), making them models for Type 2 GD. These mice show massive GlcCer accumulation in brain, liver, and lungs, and elevated proinflammatory cytokines.
4.2.2 Conditional Knockout Models
To overcome the lethality of total knockout, conditional Gba1 deletion systems have been developed:
| Model | Targeting Strategy | Phenotype | Research Applications |
|---|---|---|---|
| Gba1fl/fl; Mx1-Cre | Hematopoietic-specific deletion | Splenomegaly, cytopenia, bone disease; no CNS involvement | Type 1 GD, ERT testing, biomarker discovery |
| Gba1lnl/lnl; K14-Cre | Skin-rescued knockout | Neurodegeneration, motor dysfunction, microglial activation | Type 2/3 GD, CNS-directed therapies |
| Gba1fl/fl; Vav-Cre | Pan-hematopoietic deletion | Immune dysregulation, B-cell abnormalities | Gammopathy, malignancy risk |
| Gba1fl/fl; LysM-Cre | Myeloid-specific deletion | Splenomegaly, moderate storage | Macrophage biology, inflammation |
4.2.3 Knock-in (KI) Models
Point mutation knock-in mice better recapitulate human GD genetics:
- L444P: ~20% residual activity; neonatal lethal without skin rescue
- N370S: Variable phenotype; some models show inflammation without massive storage
- D409V: Progressive α-synuclein aggregation, memory deficits
- V394L + Saposin C null: Viable model of neuronopathic GD
4.3 Alternative Animal Models
Zebrafish (Danio rerio): gba1 knockout zebrafish develop hepatosplenomegaly and neurodegeneration, offering advantages for high-throughput drug screening and in vivo imaging.
Drosophila melanogaster: Fly models with GBA1 knockdown show lysosomal dysfunction and neurodegeneration, useful for genetic modifier screens.
5. Research Targets and Therapeutic Approaches
5.1 Enzyme Replacement Therapy (ERT)
ERT is the cornerstone of treatment for Type 1 and some Type 3 GD patients. The approach involves intravenous infusion of recombinant glucocerebrosidase that is specifically targeted to macrophages via mannose receptors.
| ERT Product | Source/Production | Dosing | Key Features |
|---|---|---|---|
| Imiglucerase (Cerezyme®) | Chinese Hamster Ovary (CHO) cells | 15-60 U/kg every 2 weeks | First approved ERT (1991); well-established safety profile |
| Velaglucerase alfa (VPRIV®) | Human fibroblast cell line | 60 U/kg every 2 weeks | Gene-activated; native carbohydrate structure |
| Taliglucerase alfa (Elelyso®) | Plant cells (carrot root) | 60 U/kg every 2 weeks | Plant-derived; no mammalian pathogens |
5.2 ERT Mechanism and Clinical Outcomes
ERT works by providing exogenous enzyme that is taken up by macrophages through mannose receptor-mediated endocytosis. Clinical benefits include:
- Hematologic improvement: Hemoglobin increases within 3-6 months; platelet counts recover in 6-12 months
- Visceral reduction: Splenic volume decreases by 30-50% within 6 months
- Skeletal stabilization: Bone pain reduction and prevention of new bone crises
- Quality of life: Reduced fatigue, improved growth in children
However, ERT limitations include inability to cross the blood-brain barrier (limiting efficacy for neuronopathic forms), high cost, and the need for lifelong intravenous infusions every two weeks.
5.3 Substrate Reduction Therapy (SRT)
SRT takes an alternative approach by inhibiting glucosylceramide synthase (GCS), the enzyme that produces GlcCer, thereby reducing substrate burden:
| SRT Agent | Mechanism | Indication | Key Considerations |
|---|---|---|---|
| Miglustat (Zavesca®) | Non-selective GCS inhibitor | Mild Type 1 GD (when ERT unavailable) | Neurological side effects; weight loss |
| Eliglustat (Cerdelga®) | Selective GCS inhibitor | Stable Type 1 GD adults | Oral administration; CYP2D6 metabolism |
| Ibiglustat | Brain-penetrant GCS inhibitor | Investigational for neuronopathic GD | Crosses blood-brain barrier; potential for CNS disease |
5.4 Pharmacological Chaperone Therapy (PCT)
PCT represents a precision medicine approach for GD patients with amenable mutations. Small molecule chaperones (e.g., ambroxol, isofagomine derivatives) bind to and stabilize mutant GCase, improving its folding and trafficking to lysosomes. This approach is mutation-specific and requires residual enzyme activity.
5.5 Gene Therapy
Gene therapy approaches under investigation include:
- Ex vivo HSC gene therapy: Lentiviral transduction of autologous hematopoietic stem cells with functional GBA1
- In vivo gene delivery: AAV vectors targeting liver or hematopoietic cells
- Genome editing: CRISPR/Cas9 correction of GBA1 mutations in patient cells
5.6 Emerging Therapeutic Targets
Novel therapeutic strategies targeting specific aspects of Gaucher disease mechanism:
| Target Pathway | Therapeutic Strategy | Development Stage |
|---|---|---|
| GCase activators | Small molecules enhancing residual enzyme activity | Preclinical/Phase I |
| Anti-inflammatory agents | C5aR1 antagonists, IL-1β inhibitors | Preclinical |
| Autophagy enhancers | TFEB activators, mTOR inhibitors | Preclinical |
| α-synuclein reduction | Antisense oligonucleotides, immunotherapy | Clinical trials (PD) |
| Calcium homeostasis | TRPML1 agonists | Preclinical |
5.7 Treatment Goals and Monitoring
Therapeutic goals in GD management include preventing irreversible complications, improving quality of life, and normalizing life expectancy. Key therapeutic targets encompass hematologic parameters (hemoglobin >11 g/dL, platelets >100,000/μL), visceral volumes (reducing spleen and liver size), skeletal health (preventing bone crises and fractures), and biomarker normalization (chitotriosidase, GlcSph levels).
- Development of brain-penetrant therapies for neuronopathic forms
- Personalized medicine approaches based on genotype and biomarker profiles
- Combination therapies (ERT + SRT + chaperones) for optimal outcomes
- Prevention strategies for GD-associated Parkinson's disease in carriers
- Gene editing as a potential curative approach
References
1. Mistry PK, et al. (2023). Refining Mouse Models of Gaucher Disease. Frontiers in Molecular Biosciences.
2. Grabowski GA. (2024). Gaucher Disease: Enzymology, Genetics, and Treatment. Annual Review of Genomics and Human Genetics.
3. Schapira AHV, et al. (2025). Classification of GBA1 variants and their impact on Parkinson's disease. npj Parkinson's Disease.
4. Barton NW, et al. (1991). Replacement therapy for inherited enzyme deficiency. New England Journal of Medicine.
5. Cox TM, et al. (2022). Enzyme Replacement or Substrate Reduction? A Review of Gaucher Disease Treatment Options. Journal of Inherited Metabolic Disease.