A Review of Recombinant β-Glucocerebrosidase in Biomedical Research
Structural Characteristics, Expression Systems, and Therapeutic Applications
Abstract
Recombinant β-glucocerebrosidase (GBA) has emerged as a cornerstone therapeutic for Gaucher disease and a critical reagent in biomedical research. This technical review systematically examines the structural characteristics, expression systems, and therapeutic applications of recombinant GBA, with particular emphasis on recent advances in protein engineering and manufacturing technologies. Comparative analysis reveals that engineered variants with optimized signal peptides demonstrate up to 5.2-fold higher expression levels, while modern purification techniques achieve monomeric enzyme purities exceeding 99%. The review further explores the expanding role of GBA in Parkinson's disease research and identifies key challenges in treating neuronopathic forms of lysosomal storage disorders.
GBA protein review, recombinant glucocerebrosidase research, enzyme replacement therapy, lysosomal storage disease, imiglucerase
Overview of β-Glucocerebrosidase (GBA)
β-Glucocerebrosidase (GBA, also known as glucosylceramidase beta or GCase) is a critical lysosomal enzyme encoded by the GBA1 gene located on chromosome 1q21. This enzyme catalyzes the hydrolysis of glucocerebroside (glucosylceramide) into glucose and ceramide, playing a pivotal role in glycosphingolipid metabolism. Deficiency or dysfunction of GBA activity leads to the accumulation of undegraded substrates within macrophage lysosomes, resulting in the pathogenesis of Gaucher disease (GD), the most prevalent lysosomal storage disorder with an estimated worldwide prevalence of 1 in 50,000 to 100,000 births.
GBA is synthesized as a precursor protein of approximately 60 kDa, which undergoes extensive post-translational modifications during trafficking to the lysosome. The enzyme exhibits optimal activity at acidic pH (4.5-5.5), consistent with its lysosomal localization. Beyond its canonical role in lipid catabolism, emerging evidence has established GBA as a significant genetic risk factor for synucleinopathies, including Parkinson's disease and dementia with Lewy bodies, highlighting its broader biomedical relevance beyond lysosomal storage disorders.
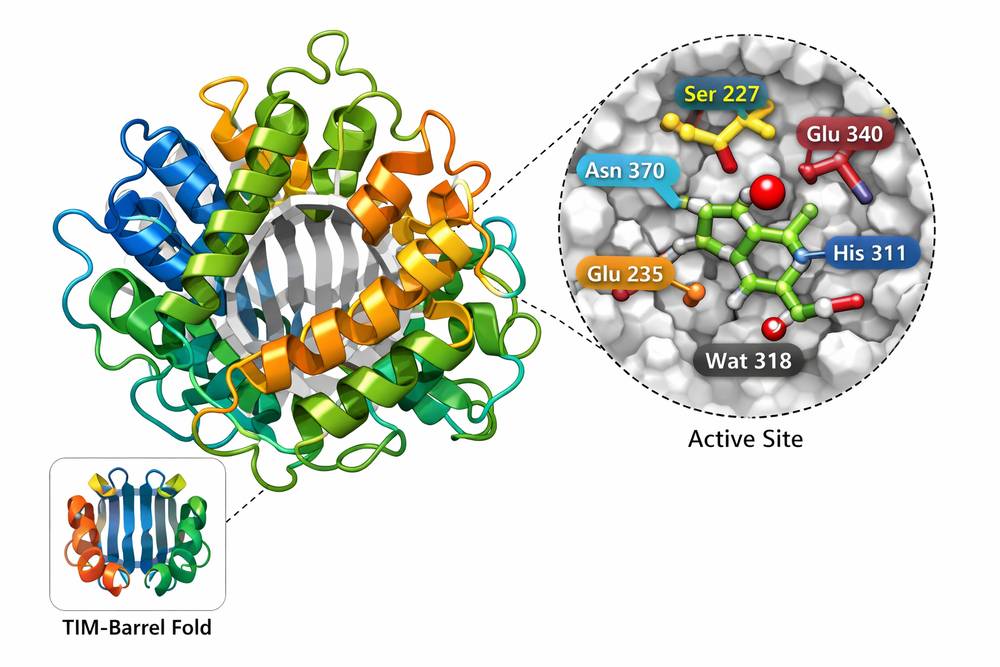
Fig 1. Molecular structure of recombinant β-glucocerebrosidase showing TIM-barrel fold and active site architecture
Structural Characteristics of Recombinant Enzyme
Molecular Architecture
The three-dimensional structure of recombinant β-glucocerebrosidase has been extensively characterized through X-ray crystallography and cryo-electron microscopy studies. The enzyme adopts a TIM-barrel fold with a distinct active site architecture that accommodates the glucocerebroside substrate.
- Catalytic Domain: The active site contains two critical glutamate residues (E340 and E235) that function as the nucleophile and acid-base catalyst, respectively.
- Glycosylation Patterns: Recombinant GBA produced in mammalian cell systems exhibits complex N-linked glycosylation at four conserved asparagine residues (N19, N59, N146, and N270). These glycan moieties are essential for proper folding, stability, and targeting to the lysosome via the mannose-6-phosphate receptor pathway.
- Signal Peptide Engineering: Recent advances have explored engineering the signal peptide with specific missense mutations derived from mammalian homologs.
Signal Peptide Engineering Performance
One engineered variant (GBA-7) containing seven amino acid substitutions in the signal peptide demonstrated 5.2-fold higher transcription levels and up to sixfold increased catalytic activity compared to wild-type controls in transient expression systems.
| Variant | Relative Transcription Level | Catalytic Activity (Fold Increase) | Expression System |
|---|---|---|---|
| Wild-type GBA | 1.0× (baseline) | 1.0× (baseline) | 293-FT cells |
| GBA-3 (3 mutations) | 2.8× | 2.1× | 293-FT cells |
| GBA-5 (5 mutations) | 4.1× | 4.5× | 293-FT cells |
| GBA-7 (7 mutations) | 5.2× | 6.0× | 293-FT cells |
Recombinant Expression Systems
Commercial Production Platforms
The production of therapeutic-grade recombinant β-glucocerebrosidase requires sophisticated expression platforms that can support proper protein folding and complex glycosylation patterns. Currently, three primary expression systems dominate the landscape:
- Chinese Hamster Ovary (CHO) Cells: The first commercially available recombinant enzyme, imiglucerase, was historically produced in CHO cells, establishing the feasibility of mammalian expression for clinical-grade GBA.
- Human Fibroblast Cell Lines: Velaglucerase alfa represents the first recombinant GBA produced in a continuous human cell line (HT-1080 fibrosarcoma cells), yielding enzyme with human-identical glycosylation patterns.
- Plant-Based Expression Systems: Taliglucerase alfa is produced in genetically engineered carrot root cells (Daucus carota), offering advantages in scalability and cost-effectiveness.
Promoter Optimization
Recent research has compared the efficacy of different promoter systems for GBA expression. The human elongation factor 1-alpha (hEF1a) promoter has demonstrated superior performance over the conventional CMV promoter in driving sustained GBA expression in 293-FT human cells.
| Promoter | Relative Expression Level | Cell Type | Optimal Application |
|---|---|---|---|
| CMV (immediate early) | 1.0× (baseline) | 293-FT | Standard transient expression |
| hEF1a (human elongation factor 1-alpha) | 2.3× | 293-FT | Sustained high-level expression |
| CAG (CMV + chicken β-actin) | 1.8× | 293-FT | Stable cell line generation |
Applications in Disease Modeling
Recombinant β-glucocerebrosidase serves as an invaluable tool in developing sophisticated disease models:
- Cellular Models: Patient-derived fibroblasts and induced pluripotent stem cells (iPSCs) from Gaucher disease patients provide platforms for testing recombinant enzyme efficacy.
- Animal Models: Genetically modified mouse models, including the Gba1 knockout and point mutation knock-in strains, serve as preclinical platforms for evaluating pharmacokinetics and therapeutic efficacy.
- Parkinson's Disease Models: Given the established genetic link between GBA mutations and Parkinson's disease risk, recombinant GBA is being explored as a potential therapeutic agent in synucleinopathy models.
Role in Gaucher Disease Research
Mechanism of Enzyme Replacement Therapy
Intravenously administered recombinant GBA is internalized by macrophages via cell surface mannose receptors. Following endocytosis and trafficking to the lysosome, the exogenous enzyme degrades accumulated glucocerebroside, thereby reversing cellular pathology and systemic manifestations.
Clinical Efficacy
Long-term studies demonstrate that ERT effectively ameliorates the visceral and hematologic manifestations of Type 1 Gaucher disease. However, the recombinant enzyme does not cross the blood-brain barrier, limiting efficacy in neuronopathic forms (Types 2 and 3).
| Parameter | Baseline (Untreated) | Post-ERT (12 months) | Post-ERT (24 months) |
|---|---|---|---|
| Liver volume (× normal) | 2.5-3.5 | 1.5-2.0 | 1.2-1.5 |
| Spleen volume (× normal) | 10-20 | 3-5 | 2-3 |
| Hemoglobin (g/dL) | 8-10 | 11-13 | 12-14 |
| Platelet count (×10⁹/L) | 50-80 | 120-150 | 150-200 |
Early administration of velaglucerase alfa at 5 months of age resulted in transient improvement of neurological symptoms, splenomegaly, and ichthyosis in Type 2 Gaucher disease, highlighting both the potential and current limitations of recombinant enzyme therapies.
Comparison with Other GBA-Based Therapeutics
The therapeutic landscape for Gaucher disease encompasses several approaches beyond recombinant enzyme replacement:
| Therapeutic Approach | Mechanism | Route | CNS Penetration | Annual Cost (USD) |
|---|---|---|---|---|
| ERT (Imiglucerase) | Exogenous recombinant GBA | IV infusion | None | $80,000-$115,000 |
| ERT (Velaglucerase) | Human-cell derived GBA | IV infusion | None | Comparable |
| ERT (Taliglucerase) | Plant-cell derived GBA | IV infusion | None | Lower |
| Substrate Reduction (Eliglustat) | Inhibit glucocerebroside synthesis | Oral | Limited | Lower |
| Pharmacological Chaperone | Stabilize mutant GBA | Oral | Potential | Moderate |
| Gene Therapy | Correction of genetic defect | Vector delivery | Potential | Procedure-dependent |
Imiglucerase remains the reference standard for ERT, with extensive long-term safety and efficacy data accumulated over three decades of clinical use.
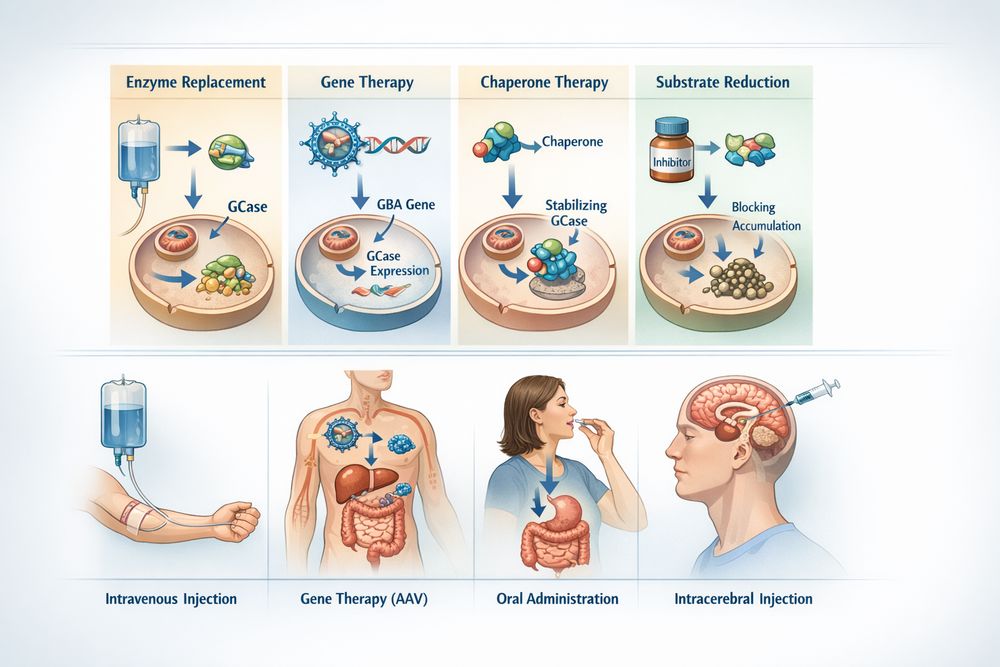
Fig 2. Comparative overview of GBA-based therapeutic mechanisms and delivery routes
Research Challenges
Despite significant advances, several challenges persist in recombinant β-glucocerebrosidase research:
- Blood-Brain Barrier Penetration: The inability of current recombinant enzymes to cross the BBB represents the most significant limitation in treating neuronopathic Gaucher disease.
- Immunogenicity: Although generally well-tolerated, some patients develop antibodies against recombinant GBA, potentially compromising therapeutic efficacy.
- Cost and Accessibility: The high annual cost of ERT poses significant barriers to global access.
- Personalized Medicine: The vast allelic heterogeneity of Gaucher disease (over 400 documented GBA mutations) necessitates personalized therapeutic approaches.
Next-Generation Engineering Targets
| Engineering Target | Current Status | Potential Impact |
|---|---|---|
| Signal peptide optimization | Validated (GBA-7) | 5× production increase |
| Glycosylation engineering | In development | Enhanced macrophage uptake |
| Fusion with BBB shuttle peptides | Preclinical | CNS delivery capability |
| Catalytic efficiency enhancement | Computational design | Reduced dosing frequency |
Conclusion
Recombinant β-glucocerebrosidase represents a triumph of biotechnology in transforming a once-fatal lysosomal storage disorder into a manageable chronic condition. From the pioneering development of imiglucerase to current innovations in protein engineering and expression technologies, this field continues to evolve.
Core Quality Standards for Research-Grade Recombinant GBA:
- Purity: SEC-HPLC monomer content ≥99%
- Specific Activity: ≥200 U/mg protein
- Glycosylation Profile: Terminal mannose residues for macrophage targeting
- Endotoxin Level: <0.1 EU/μg
- Stability: 24-month shelf life at 2-8°C
Future research must address the unmet needs in neuronopathic disease, improve treatment accessibility, and leverage emerging technologies for next-generation therapeutics. The expanding role of GBA in Parkinson's disease and other synucleinopathies further underscores the importance of continued investment in recombinant enzyme research and development.
For precision medicine research, selecting high-quality recombinant GBA products with documented manufacturing processes and comprehensive quality data is essential. We encourage suppliers to provide detailed certificates of analysis and transparency regarding expression systems to advance lysosomal disease research standardization.
References
1. Brady, R.O., et al. (1965). N Engl J Med, 273: 1163-1167.
2. Engineering synthetic and recombinant human lysosomal β-glucocerebrosidase for enzyme replacement therapy. SN Applied Sciences, 2024, 6: 622.
3. Grabowski, G.A., et al. (2015). Lancet, 386(9986): 443-453.
4. Drug Class Review: Targeted Therapies for Gaucher Disease. Oregon Health & Science University, 2019.
5. Sidransky, E., et al. (2009). N Engl J Med, 361: 1651-1661.
6. Mistry, P.K., et al. (2015). Am J Hematol, 90(S1): S27-S35.
7. Takajo, D., et al. (2020). Iran J Pediatr, 30(2): e98996.