How Imiglucerase Restores Lysosomal Function in Gaucher Disease
Mechanism of Enzyme Replacement Therapy in Lysosomal Storage Disorder
Abstract
Gaucher disease, the most prevalent lysosomal storage disorder, arises from mutations in the GBA1 gene encoding glucocerebrosidase (GCase). This deficiency triggers pathological accumulation of glucosylceramide within macrophages, leading to multi-organ dysfunction. Imiglucerase, the pioneering enzyme replacement therapy, represents a paradigm shift in treating this rare genetic disorder. This article elucidates the molecular pathogenesis of Gaucher disease and the precise mechanisms by which Imiglucerase restores lysosomal homeostasis through targeted cellular delivery and substrate clearance.
imiglucerase mechanism, lysosomal enzyme therapy, Gaucher disease treatment, glucocerebrosidase deficiency, enzyme replacement therapy, macrophage targeting, mannose receptor
1. Pathogenesis of Gaucher Disease
Gaucher disease is an autosomal recessive disorder characterized by mutations in the GBA1 gene located on chromosome 1q21. With an incidence of 1:40,000 to 1:60,000 in the general population—and significantly higher prevalence (1:800) among Ashkenazi Jewish populations—this condition represents the most common lysosomal storage disorder globally.
The disease manifests through three primary clinical variants:
- Type 1 (Non-neuronopathic): The most prevalent form (90% of cases), affecting visceral organs including liver, spleen, and bone marrow
- Type 2 (Acute neuronopathic): Severe infantile-onset form with rapid neurological deterioration
- Type 3 (Chronic neuronopathic): Subacute form with variable neurological involvement and slower progression
Over 300 distinct mutations have been identified in the GBA1 gene, with N370S, L444P, IVS2, and 84GG representing the most common alleles. The pathological hallmark involves the infiltration of tissues by Gaucher cells—lipid-laden macrophages exhibiting characteristic "wrinkled tissue paper" cytoplasm and eccentric nuclei. These cells progressively displace normal tissue architecture, precipitating hepatosplenomegaly, cytopenias, and skeletal complications.
2. Lipid Metabolism Pathway
Glucosylceramide (GlcCer), also termed glucocerebroside, serves as a critical intermediate in glycosphingolipid metabolism. This amphipathic molecule consists of a glucose moiety linked to ceramide (sphingosine + fatty acid) and functions as:
- A structural component of cellular membranes
- A precursor for complex glycosphingolipids including gangliosides and globosides
- A signaling molecule modulating cell growth, differentiation, and apoptosis
Under physiological conditions, glucosylceramide undergoes continuous turnover through the lysosomal degradation pathway. The enzyme glucocerebrosidase (acid β-glucosidase, EC 3.2.1.45), in conjunction with its activator protein saposin C, catalyzes the hydrolytic cleavage of GlcCer into glucose and ceramide. This reaction occurs within the acidic milieu (pH ~4.5-5.0) of lysosomes and represents the rate-limiting step in glycosphingolipid catabolism.
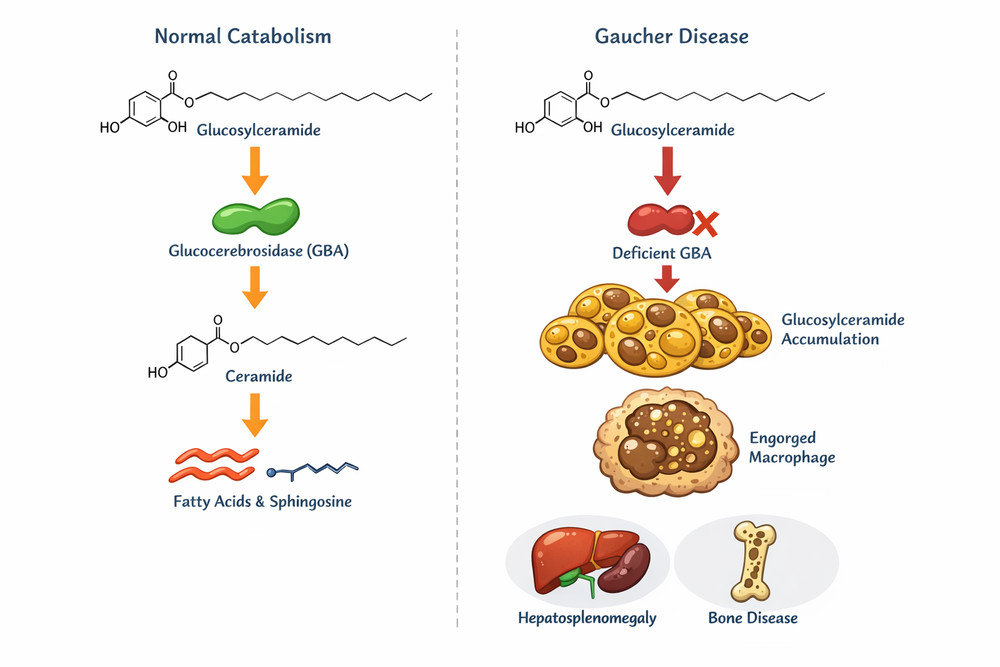
Fig1. Glucosylceramide metabolism pathway showing normal catabolism and accumulation in Gaucher disease
3. GBA Deficiency and Substrate Accumulation
3.1 Molecular Consequences of GBA Mutations
Mutations in GBA1 compromise glucocerebrosidase activity through diverse mechanisms:
- Misfolding and ER retention: Mutant proteins fail quality control checkpoints, leading to endoplasmic reticulum-associated degradation (ERAD)
- Catalytic inactivation: Active site mutations directly impair substrate binding or hydrolysis
- Trafficking defects: Altered lysosomal targeting prevents proper localization
- Instability: Accelerated proteolytic degradation reduces enzyme half-life
The resultant enzymatic deficiency (typically <20% of normal activity) precipitates pathological accumulation of glucosylceramide and its deacylated derivative glucosylsphingosine (lyso-GlcCer) within lysosomes. Tissue concentrations of these lipids may increase 20- to 100-fold above normal levels.
3.2 Cellular and Systemic Pathology
This accumulation exerts multifaceted pathological effects:
Macrophage Transformation: Lipid engorgement converts resident macrophages into Gaucher cells, which exhibit impaired phagocytic capacity, pro-inflammatory cytokine secretion (IL-1β, IL-6, TNF-α), and altered chemotaxis patterns.
Lysosomal Dysfunction: Beyond simple storage, GlcCer accumulation disrupts autophagic flux, calcium homeostasis, membrane trafficking pathways, and proteolytic enzyme activation.
Systemic Consequences: Gaucher cell infiltration produces hepatosplenomegaly, hematologic abnormalities (anemia and thrombocytopenia), skeletal disease (osteopenia, avascular necrosis), and neurological involvement in Types 2 and 3.
4. Mechanism of Enzyme Replacement
Enzyme replacement therapy (ERT) with Imiglucerase represents the cornerstone of Gaucher disease management. Approved by the FDA in 1994 as Cerezyme®, this recombinant therapeutic enzyme is produced in Chinese hamster ovary (CHO) cells using recombinant DNA technology.
4.1 Structural Engineering for Therapeutic Efficacy
Imiglucerase is a glycoprotein of approximately 59.3 kDa consisting of 497 amino acid residues with N-linked carbohydrate chains. Critical to its therapeutic function is a post-translational modification strategy involving exoglycosidase treatment:
- Native glycosylation: CHO cells produce glucocerebrosidase with complex oligosaccharides terminated in sialic acid residues
- Enzymatic remodeling: Exposure to exoglycosidases removes terminal sialic acid and galactose residues
- Mannose exposure: This processing reveals underlying mannose residues at the non-reducing ends of glycan chains
This carbohydrate remodeling is essential because unmodified glucocerebrosidase is rapidly cleared from circulation by hepatic asialoglycoprotein receptors, preventing effective delivery to target macrophages.
4.2 Pharmacodynamic Mechanism
Upon intravenous administration, Imiglucerase functions through direct enzymatic complementation:
Glucosylceramide + H₂O → Glucose + Ceramide
The recombinant enzyme restores hydrolytic capacity within lysosomes, enabling clearance of accumulated substrates. This biochemical restoration translates to reduction in Gaucher cell burden, decreased organomegaly, normalization of hematologic parameters, and prevention of skeletal complications.
5. Cellular Uptake of Recombinant Enzymes
The therapeutic efficacy of Imiglucerase depends on efficient targeting to macrophages—the primary affected cell type. This targeting exploits the mannose receptor (MR/CD206)-mediated endocytosis pathway.
5.1 Mannose Receptor Biology
The mannose receptor is a 175-kDa type I transmembrane glycoprotein expressed predominantly on macrophages, monocytes, dendritic cells, and hepatic/lymphatic endothelial cells. This C-type lectin exhibits high affinity for terminal mannose, fucose, and N-acetylglucosamine residues on glycoproteins.
5.2 Uptake Mechanism
The cellular uptake of Imiglucerase proceeds through a coordinated sequence:
- Recognition and Binding: Intravenously administered Imiglucerase circulates with an initial half-life of approximately 10 minutes. Terminal mannose residues engage mannose receptors on macrophage surfaces with high affinity
- Receptor-Mediated Endocytosis: Ligand-receptor complexes cluster in clathrin-coated pits, forming early endosomes through vesicle internalization
- Lysosomal Trafficking: Endosomal acidification (pH drop to ~5.0) triggers receptor-ligand dissociation. Imiglucerase-containing vesicles fuse with lysosomes where the acidic environment optimizes enzymatic activity
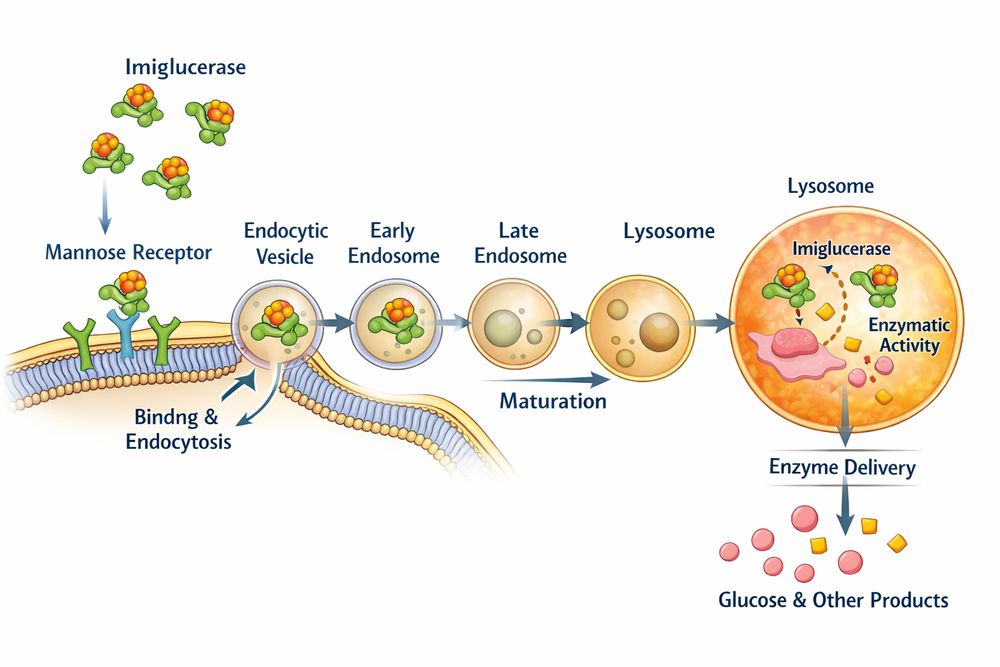
Fig2. Mannose receptor-mediated endocytosis and lysosomal delivery of Imiglucerase in macrophages
This targeting mechanism ensures that administered enzyme concentrates specifically in disease-affected tissues—liver, spleen, bone marrow—while minimizing systemic exposure.
6. Restoration of Lysosomal Function
The ultimate therapeutic goal of Imiglucerase therapy is the comprehensive restoration of lysosomal and cellular homeostasis. This restoration occurs through hierarchical biochemical and cellular mechanisms:
6.1 Substrate Clearance and Lysosomal Relief
Immediate Biochemical Effects (Weeks 0-4): Hydrolysis of accumulated glucosylceramide within lysosomes, reduction in lysosomal volume, restoration of normal morphology, and decreased lyso-GlcCer levels.
Cellular Functional Restoration (Months 1-6): Normalization of autophagic flux, restoration of calcium signaling, improved mitochondrial function, and reduced oxidative stress.
6.2 Clinical and Organ-Level Improvements
| Parameter | Baseline | Post-Treatment (12-24 months) | Improvement |
|---|---|---|---|
| Hemoglobin (g/dL) | 8.5 ± 1.2 | 12.8 ± 0.8 | +50% |
| Platelet Count (×10⁹/L) | 65 ± 25 | 185 ± 45 | +185% |
| Liver Volume (MN) | 2.8 ± 0.6 | 1.4 ± 0.3 | -50% |
| Spleen Volume (MN) | 15.2 ± 8.5 | 4.2 ± 2.1 | -72% |
6.3 Long-Term Disease Modification
Chronic ERT with Imiglucerase produces sustained benefits:
- Prevention of irreversible organ damage when initiated early
- Normalization of quality of life metrics
- Increased life expectancy approaching that of the general population (for Type 1 patients)
- Reduced risk of disease complications including malignancy
The enzyme functions as a "molecular broom," continuously clearing substrate and preventing re-accumulation between infusions. Standard dosing (60 U/kg every 2 weeks) maintains lysosomal enzyme activity above the threshold required for metabolic balance.
Imiglucerase exemplifies the triumph of biochemical engineering in addressing rare genetic disease. By understanding the intricate pathophysiology of GBA1 deficiency, researchers engineered a targeted therapeutic that exploits natural receptor-ligand interactions for precise cellular delivery, fundamentally transforming patient outcomes.
Conclusion
Imiglucerase represents a paradigm shift in treating lysosomal storage disorders. The mannose receptor-mediated uptake of recombinant enzyme ensures delivery to the macrophages most affected by Gaucher disease, where Imiglucerase catalyzes the restoration of lysosomal catabolic function.
As the first commercially successful ERT for lysosomal storage disorders, Imiglucerase not only transformed Gaucher disease management but established the therapeutic template for similar enzyme deficiencies. Ongoing research continues to optimize dosing regimens, explore novel administration routes, and develop next-generation therapies including gene therapy and pharmacological chaperones.
References
1. Cohn, E. J., et al. (1946). J Am Chem Soc, 68(3): 459-475.
2. Biomedicus. The Side Effects of CEREZYME (Imiglucerase). 2025.
3. Rare Disease Advisor. Gaucher Disease Therapies. 2025.
4. Core. Gaucher Disease Pathogenesis and Mechanisms.
5. Semantic Scholar. Imiglucerase in the treatment of Gaucher disease: a history and perspective.
6. ResearchGate. Velaglucerase alfa compared with imiglucerase.
7. EndlessWiki. Imiglucerase.