Frequently Asked Questions About Imiglucerase in Research
Essential Information for Researchers Working with Recombinant Glucocerebrosidase
1. What is Recombinant Imiglucerase?
Imiglucerase is a recombinant DNA-derived form of human glucocerebrosidase (GBA, EC 3.2.1.45), the lysosomal enzyme deficient in Gaucher disease. Produced in Chinese hamster ovary (CHO) cells, this therapeutic protein catalyzes the hydrolysis of glucocerebroside to glucose and ceramide, correcting the metabolic defect underlying the disease pathology.
1.1 Molecular Characteristics
The recombinant enzyme consists of 497 amino acids with a molecular weight of approximately 60.8 kDa. Three N-linked glycosylation sites (Asn19, Asn59, Asn146) are modified with complex oligosaccharides terminating in mannose residues, facilitating uptake by macrophage mannose receptors. This glycosylation pattern differs from native placental-derived enzyme, optimizing tissue targeting.
1.2 Manufacturing and Quality Attributes
Production occurs in serum-free CHO cell culture under current Good Manufacturing Practice (cGMP) conditions. The purification process includes multiple chromatographic steps, viral inactivation, and extensive quality testing. Each batch undergoes analysis for enzyme activity, purity, endotoxin levels, and bioburden to ensure consistency and safety.
Research-grade imiglucerase provides high specific activity (>10 units/mg protein) suitable for in vitro studies, cell culture applications, and biochemical assays. The product is available in multiple formulations to match diverse experimental requirements.
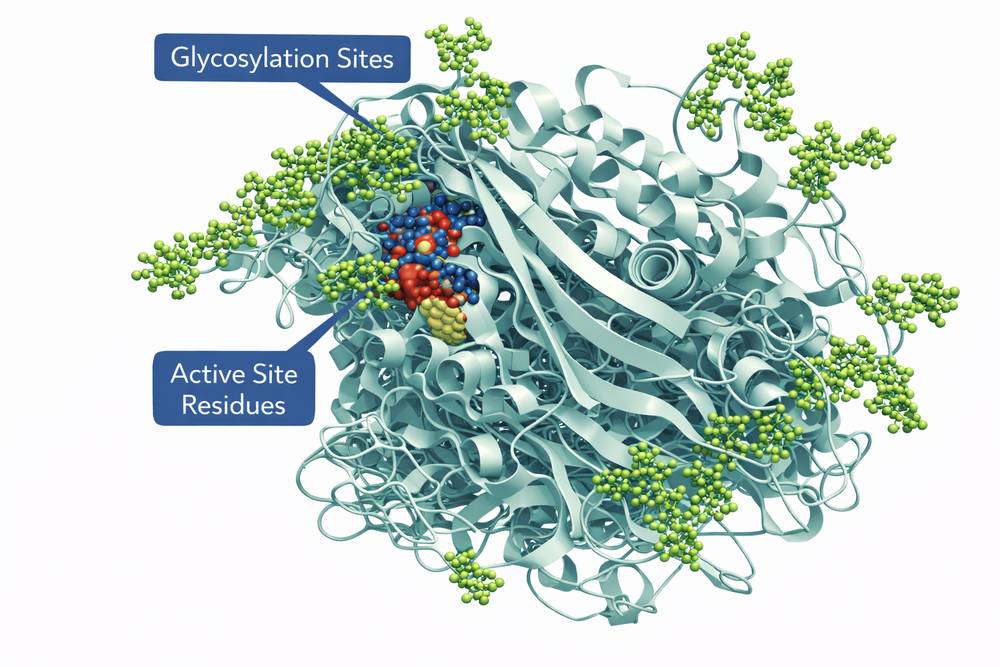
Fig 1. Molecular structure of recombinant imiglucerase showing glycosylation sites and active site residues
2. What Research Fields Use Imiglucerase?
Imiglucerase serves as a critical reagent across multiple biomedical research disciplines. Its applications extend beyond Gaucher disease research into broader areas of lysosomal biology, neurodegeneration, and enzyme engineering.
2.1 Lysosomal Storage Disease Research
Primary applications include studies of Gaucher disease pathophysiology, enzyme kinetics, and substrate reduction strategies. Researchers utilize imiglucerase to establish cellular models of enzyme replacement therapy, investigate uptake mechanisms in different cell types, and evaluate the impact of glycosylation variants on tissue distribution.
2.2 Parkinson's Disease and Neurodegeneration
The discovery that GBA mutations represent the strongest genetic risk factor for Parkinson's disease has expanded imiglucerase applications into neurodegeneration research. Studies employ the enzyme to investigate lysosomal dysfunction in dopaminergic neurons, examine α-synuclein aggregation mechanisms, and test whether enzyme augmentation can mitigate neurodegenerative processes.
2.3 Enzyme Engineering and Drug Development
Protein engineering studies utilize imiglucerase as a template for developing improved variants with enhanced stability, brain penetration, or targeting specificity. Researchers modify glycosylation patterns, engineer fusion proteins, or develop alternative delivery systems. These studies contribute to next-generation therapies for both Gaucher disease and related conditions.
| Research Area | Specific Applications | Typical Assay Formats | Key Considerations |
|---|---|---|---|
| Gaucher Disease | ERT mechanism studies; Cellular uptake | Cell culture; Enzyme assays | Macrophage targeting essential |
| Parkinson's Research | Neuroprotection; Autophagy modulation | Neuronal cultures; Animal models | CNS delivery limitations |
| Enzyme Kinetics | Catalytic mechanism; Inhibitor screening | Spectrophotometric assays | Saposin C cofactor required |
| Protein Chemistry | Glycosylation analysis; Stability studies | Mass spectrometry; DSC | Glycoform heterogeneity |
| Drug Delivery | Nanoparticle encapsulation; Brain targeting | Pharmacokinetic studies | Blood-brain barrier penetration |
3. How is Enzyme Activity Measured?
Accurate quantification of imiglucerase activity is essential for quality control, dosing calculations, and research applications. Multiple assay formats are available, each with distinct advantages depending on the experimental context.
3.1 Fluorometric Assay (Standard Method)
The most widely used method employs 4-methylumbelliferyl-β-D-glucopyranoside (4-MUG) as artificial substrate. Upon hydrolysis, the fluorescent product 4-methylumbelliferone (4-MU) is released and quantified. The reaction requires sodium taurocholate as detergent and sodium citrate buffer at pH 5.5-6.0. One unit of activity is defined as the amount of enzyme that hydrolyzes 1 μmol of substrate per minute under standard conditions.
3.2 Natural Substrate Assay
For physiologically relevant measurements, assays utilizing natural substrate glucocerebroside are employed. These methods typically use radiolabeled substrate (C14-glucocerebroside) or mass spectrometry detection. While more complex than fluorometric assays, natural substrate methods provide accurate assessment of catalytic efficiency toward the physiological substrate and are essential for regulatory submissions.
3.3 High-Throughput Screening Formats
Research applications often require rapid activity assessment. Modified fluorometric assays compatible with 96-well or 384-well plates enable screening of large compound libraries for inhibitors or activators. These formats maintain sensitivity while reducing reagent consumption and assay time. Critical parameters include reaction linearity, substrate concentration optimization, and proper controls for background fluorescence.
Always include saposin C (cofactor) when measuring activity against natural substrate. For 4-MUG assays, ensure pH is strictly maintained at 5.5-5.8 as activity drops significantly outside this range. Pre-incubate enzyme at assay temperature for 5 minutes before substrate addition.
| Assay Method | Substrate | Detection | Sensitivity | Applications |
|---|---|---|---|---|
| Fluorometric (4-MUG) | 4-Methylumbelliferyl-glucoside | Fluorescence (365/450 nm) | High (0.01 U/mL) | QC; Routine research |
| Natural Substrate | Glucocerebroside | Radioactivity or LC-MS | Moderate (0.1 U/mL) | Regulatory; Kinetic studies |
| Colorimetric | pNPG (p-nitrophenyl-glucoside) | Absorbance (405 nm) | Moderate (0.05 U/mL) | Teaching; Quick screening |
| Coupled Assay | Glucose oxidase coupling | Absorbance or fluorescence | High | Continuous monitoring |
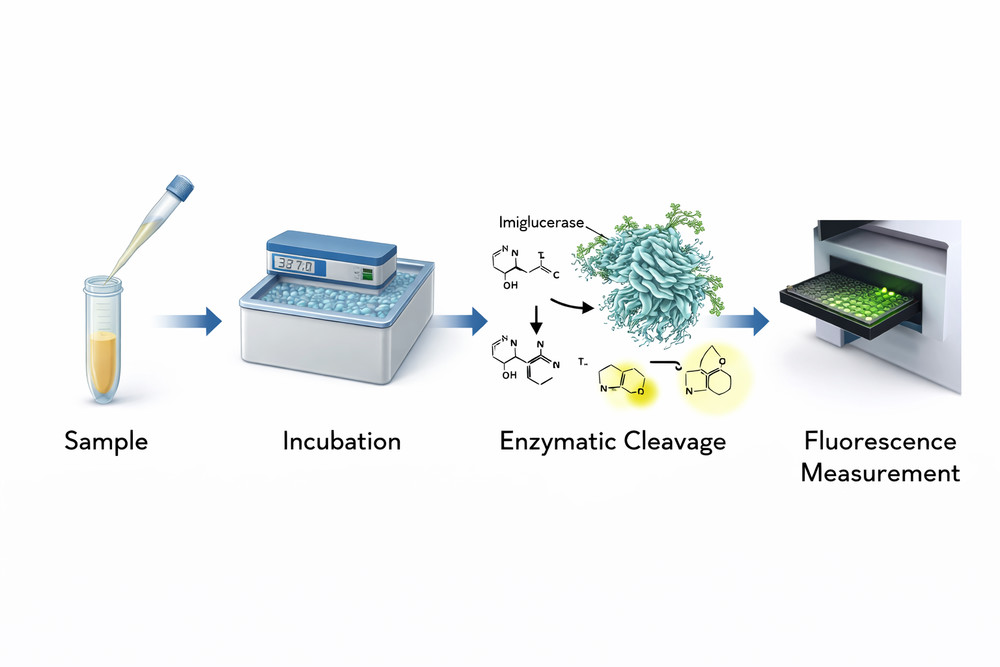
Fig 2. Workflow for imiglucerase activity measurement using fluorometric substrate
4. What Models Are Suitable for Imiglucerase Research?
Selection of appropriate experimental models depends on the research question, available resources, and translational relevance. Models range from simple biochemical systems to complex animal models, each offering distinct advantages.
4.1 Cellular Models
Patient-derived fibroblasts from Gaucher disease patients provide authentic disease backgrounds with endogenous substrate accumulation. These cells are particularly valuable for studying enzyme uptake, lysosomal trafficking, and cellular correction. Macrophage cell lines (THP-1, RAW264.7) model the primary target cells for ERT and enable high-throughput screening. Induced pluripotent stem cell (iPSC)-derived macrophages and neurons offer genetically defined systems for mechanistic studies.
4.2 Biochemical and Biophysical Systems
Purified enzyme preparations allow detailed kinetic characterization, including Km and kcat determinations, inhibition constant measurements, and stability assessments. Liposome systems reconstitute the membrane environment, enabling studies of lipid substrate presentation and saposin C cofactor function. These reductionist approaches provide fundamental insights into enzyme mechanism.
4.3 Animal Models
Gba knockout mice recapitulate severe Type 2 Gaucher disease but require genetic manipulation for viability. Conditional knockout models enable tissue-specific deficiency. Gba hypomorphic mice (D409V/D409V) model neuronopathic disease with longer survival. These models are essential for pharmacokinetic studies, tissue distribution analysis, and preclinical efficacy testing of novel formulations.
| Model System | Key Features | Applications | Limitations |
|---|---|---|---|
| Patient Fibroblasts | Authentic disease background; Human genetic context | Uptake studies; Correction assays | Limited availability; Variability |
| THP-1 Macrophages | Homogeneous population; Scalable | High-throughput screening; Uptake kinetics | Transformed cell line artifacts |
| iPSC-Derived Cells | Isogenic controls; Disease-specific mutations | Neurodegeneration studies; Personalized models | Complex differentiation; Cost |
| Gba Knockout Mice | Complete deficiency; Severe phenotype | Pathophysiology; Early lethality rescue | Perinatal lethal; Requires intervention |
| Gba Hypomorphic Mice | Residual activity; Neuronopathic features | Drug testing; Longitudinal studies | Variable penetrance; Short lifespan |
5. Storage and Stability Considerations
Proper handling and storage are critical for maintaining imiglucerase activity and preventing degradation. The enzyme is susceptible to denaturation, aggregation, and loss of glycosylation integrity under inappropriate conditions.
5.1 Recommended Storage Conditions
Lyophilized powder should be stored at -20°C or -80°C in airtight containers with desiccant. Under these conditions, the product remains stable for 24 months or longer. Reconstituted enzyme is stable for 1-2 weeks at 4°C, or 6 months at -80°C when stored in aliquots to avoid freeze-thaw cycles. Repeated freezing and thawing causes aggregation and activity loss.
5.2 Formulation and Buffer Considerations
Reconstitution in neutral pH buffers (PBS, HEPES) maintains stability. Avoid extreme pH (<5.0 or >8.0) which promotes denaturation. Addition of stabilizers such as BSA (0.1-1%) or glycerol (10-20%) can enhance stability for dilute solutions. The presence of reducing agents (DTT, β-mercaptoethanol) should be minimized as they can affect disulfide bond integrity.
5.3 Activity Monitoring and Quality Control
Establish baseline activity upon receipt and monitor periodically during storage. Significant activity loss (>10%) indicates degradation requiring fresh preparation. Visual inspection for precipitation or turbidity provides rapid assessment of aggregation. For critical experiments, always verify activity immediately before use.
Store research-grade imiglucerase at -80°C in single-use aliquots to prevent freeze-thaw damage. Reconstitute immediately before use in sterile, neutral pH buffer. Discard if visible precipitation or >10% activity loss is observed.
| Storage Format | Temperature | Stability Period | Special Considerations |
|---|---|---|---|
| Lyophilized powder | -20°C to -80°C | 24+ months | Protect from moisture; Desiccant recommended |
| Concentrated solution (1 mg/mL) | -80°C (single aliquots) | 6 months | No freeze-thaw cycles; Snap freeze in liquid N2 |
| Working solution (0.1 mg/mL) | 4°C | 1-2 weeks | Add stabilizer (BSA 0.1%); Monitor for precipitation |
| Dilute solution (<0.01 mg/mL) | 4°C (immediate use) | 24 hours | Adsorption losses; Use siliconized tubes |
| Room temperature | 20-25°C | 8 hours maximum | Activity loss 5-10% per day; Avoid if possible |
Conclusion
Imiglucerase represents a cornerstone reagent for lysosomal disease research, with applications extending from basic biochemistry to translational medicine. Understanding the molecular properties, assay requirements, and handling procedures ensures optimal experimental outcomes. As research expands into neurodegeneration and enzyme engineering, the demand for high-quality, well-characterized imiglucerase preparations continues to grow.
Researchers should select appropriate models and assays based on specific experimental questions, always considering the physiological context of enzyme function. Proper storage and handling preserve activity and ensure reproducibility across experiments. For additional technical support or product inquiries, our scientific team remains available to assist with protocol optimization and troubleshooting.
References
1. Barton, N. W., et al. (1991). N Engl J Med, 324(21): 1464-1470.
2. Grabowski, G. A. (2012). Ann Intern Med, 156(1 Pt 1): 55-62.
3. Beutler, E., & Gelbart, T. (1996). Proc Natl Acad Sci USA, 93(18): 9547-9550.
4. Mistry, P. K., et al. (2017). Am J Hematol, 92(2): 190-198.