Mechanistic Diversity of IVIg in Murine Autoimmune Models: From Fc Receptor Saturation to Cytokine Modulation
Comprehensive Analysis of Fc-Dependent and Independent Pathways
Abstract
Intravenous immunoglobulin (IVIg), as a pleiotropic immunomodulatory therapy, exhibits a sophisticated network of mechanisms in autoimmune disease treatment. Contrary to conventional understanding, IVIg's therapeutic effects do not stem from a single molecular event but rather from multi-level, multi-target synergistic actions that reshape immune homeostasis. This review systematically examines the four core mechanisms of IVIg in autoimmune disease animal models: Fc receptor blockade, autoantibody neutralization, complement regulation, and dendritic cell functional modulation. We focus on delineating the molecular distinctions and spatiotemporal dynamics between FcγRIIB-dependent pathways and sialylation-mediated Fc-independent pathways. By integrating key experimental data from ITP, EAE, and SLE mouse models, we elucidate the molecular basis and clinical translation potential of IVIg's in vivo immunomodulation. Notably, the contribution weight of different mechanisms varies significantly across disease progression, providing a theoretical framework for developing next-generation engineered IVIg formulations.
IVIg mechanism of action, Fc receptor blockade, ITP mouse model, experimental autoimmune encephalomyelitis (EAE), immunomodulation in vivo, IVIg products
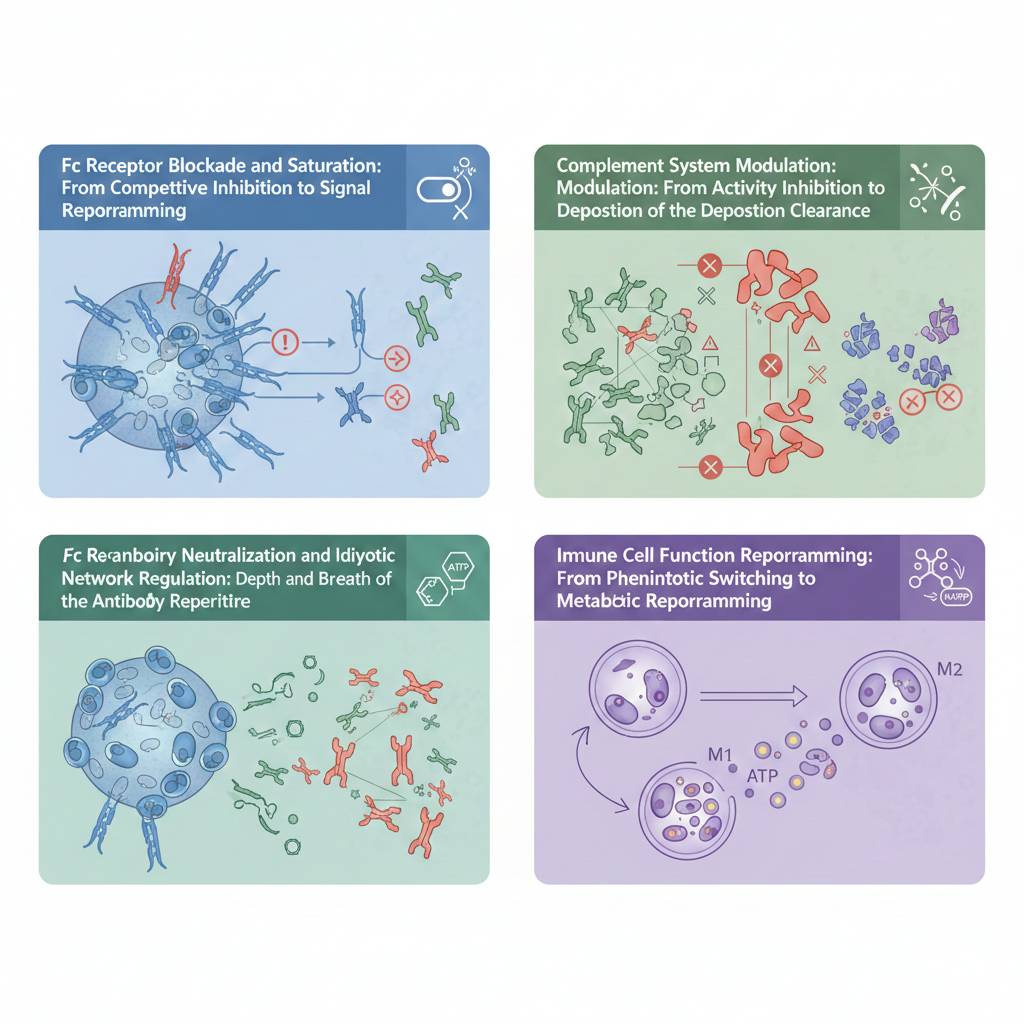
Fig 1. Schematic overview of the four major mechanistic pathways of IVIg in autoimmune disease models
1. Mechanistic Overview: The Four Pathways and Their Synergistic Network
1.1 Historical Background and Evolution of Mechanistic Understanding
Intravenous immunoglobulin (IVIg), a broad-spectrum IgG mixture extracted from healthy donor plasma, exerts therapeutic effects in autoimmune diseases far beyond simple antibody replacement or neutralization. Since its first successful use in idiopathic thrombocytopenic purpura (ITP) treatment in 1981, research on IVIg's mechanisms of action has undergone three important paradigm shifts: from initial Fc receptor blockade theory, to the discovery of FcγRIIB upregulation in the early 21st century, to the recent elucidation of sialylation-mediated Fc-independent pathways. This deepening understanding reveals the essence of IVIg as an "immunomodulatory cocktail"—containing multiple functional components that exert differential effects in various disease microenvironments.
1.2 Fc Receptor Blockade and Saturation: From Competitive Inhibition to Signal Reprogramming
IVIg saturates Fcγ receptors (FcγRs) through high-concentration IgG Fc fragments, with the concentration dependence being particularly critical. At therapeutic doses (typically 1-2 g/kg), serum IgG concentrations can reach 5-10 times normal levels, creating a competitive advantage. This saturation effect is dual in nature: on one hand, it competitively inhibits binding of pathogenic immune complexes to activating FcγRs (such as FcγRI and FcγRIII), blocking downstream ITAM phosphorylation cascades; on the other hand, IVIg specifically upregulates expression of the inhibitory receptor FcγRIIB, reshaping immune cell activation thresholds. This "dual regulatory" pattern is particularly prominent in macrophages and dendritic cells, shifting cellular responses to immune complexes from activation to inhibition. Notably, FcγRIIB upregulation involves not merely transcriptional changes but epigenetic modifications such as histone acetylation, leading to sustained gene expression.
1.3 Autoantibody Neutralization and Idiotypic Network Regulation: Depth and Breadth of the Antibody Repertoire
IVIg contains millions of unique antibody variable regions, forming a vast "antibody pool." This diversity enables it to neutralize pathogenic autoantibodies through anti-idiotypic antibodies or block their antigen-binding sites. In SLE models, IVIg's anti-dsDNA idiotypic antibodies can recognize and neutralize pathogenic anti-dsDNA antibodies, reducing their deposition in kidneys. A deeper effect involves IVIg's ability to reshape B cell receptor (BCR) signaling by cross-linking BCR with FcγRIIB, inducing B cell anergy or apoptosis. This process is particularly critical in germinal center reactions, effectively suppressing affinity maturation of autoreactive B cells.
1.4 Complement System Modulation: From Activity Inhibition to Deposition Clearance
IVIg's regulation of the complement system exhibits multi-target characteristics. First, IVIg can sequester complement activation products C3b/C4b, interfering with classical pathway initiation through its Fc region's C1q-binding site, thereby reducing C3 convertase formation. Second, IVIg accelerates complement dissolution, promotes C3b degradation, and inhibits membrane attack complex (MAC) deposition on target cells, protecting organs from complement-mediated damage. In ITP models, IVIg treatment reduces platelet surface C3d deposition by over 70%, an effect that synergizes with its anti-phagocytic action to significantly enhance platelet survival. Additionally, IVIg can upregulate expression of complement regulatory proteins (such as CD55 and CD59) on blood cells and endothelial cells, enhancing endogenous complement inhibition.
1.5 Immune Cell Function Reprogramming: From Phenotypic Switching to Metabolic Reprogramming
IVIg directly regulates dendritic cells (DCs), macrophages, and B cells. In DCs, IVIg inhibits secretion of pro-inflammatory cytokines (TNF-α, IL-6, IL-12), promotes production of anti-inflammatory mediators (IL-10, TGF-β), and induces a tolerogenic DC phenotype characterized by low co-stimulatory molecule (CD80/CD86) expression and high inhibitory molecule (PD-L1) expression. This phenotypic switching is closely associated with metabolic reprogramming: IVIg drives DCs from glycolysis toward oxidative phosphorylation, reducing mTOR activity and thereby suppressing inflammatory responses. In macrophages, IVIg drives M1-to-M2 polarization, enhances scavenger receptor expression, and promotes apoptotic cell clearance. Regulation of T cell subsets manifests as inhibiting Th1/Th17 differentiation while promoting Treg expansion, partially mediated by IL-27 and TGF-β secreted from DCs.
IVIg's therapeutic effect relies on a "dual regulatory" mode: blocking activating FcγRs while upregulating inhibitory FcγRIIB, fundamentally resetting immune cell activation thresholds.
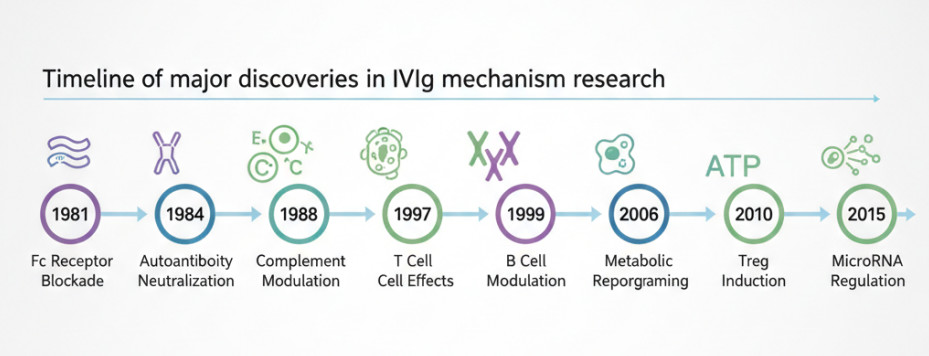
Fig 2. Timeline of major discoveries in IVIg mechanism research
2. Fc Receptor-Dependent Mechanisms: Core Role of FcγRIIB in ITP Models and Signal Transduction Analysis
2.1 ITP Mouse Model Establishment and Phenotypic Validation of IVIg Efficacy
In passive ITP models, injection of anti-platelet monoclonal antibodies (such as anti-CD41 IgG1 or IgG2a) induces acute thrombocytopenia, mimicking the core pathological feature of human ITP: antibody-mediated macrophage phagocytosis. This model offers rapid induction (platelets drop below 20% of baseline within 24 hours) and strong reversibility, providing an ideal platform for studying IVIg's acute effects. Studies show that IVIg treatment (1-2 g/kg) can significantly increase platelet counts to 60-80% of baseline within 24 hours. This effect is prominent in wild-type (WT) mice but completely absent in *Fcgr2b* knockout mice, indicating that FcγRIIB is an essential effector molecule. Notably, low-dose IVIg (0.5 g/kg) shows limited efficacy in WT mice but remains significantly effective in *Fcgr2b* overexpression mice, further confirming the quantitative dose-receptor expression relationship.
2.2 Mechanistic Analysis: SH2 Domains and Phosphoinositide Signaling Cascades
The intracellular domain of FcγRIIB contains an immunoreceptor tyrosine-based inhibitory motif (ITIM). Upon IVIg stimulation, ITIM phosphorylation recruits the SH2 domain-containing inositol phosphatase SHIP-1. Once recruited, SHIP-1 hydrolyzes PIP3 to PIP2, thereby blocking sustained PI3K/Akt pathway activation—the core molecular event inhibiting macrophage phagocytosis of platelets. Studies demonstrate that IVIg treatment upregulates FcγRIIB expression 2-3 fold in splenic marginal zone macrophages, accompanied by a 4-5 fold enhancement in SHIP-1 phosphorylation. This enhancement is cell-specific: changes are not significant in red pulp macrophages but particularly prominent in marginal zone macrophages, consistent with the latter's dominant role in IVIg-mediated platelet clearance. Additionally, IVIg can upregulate other inhibitory molecules such as IRAK-M and SOCS1, forming a multi-layered negative regulatory network.
2.3 Cell-Specific Regulation and Spatiotemporal Dynamics
Recent studies using conditional knockout mice reveal that FcγRIIB expressed on dendritic cells is crucial for IVIg's long-term immunomodulation, not just macrophages. DC-specific *Fcgr2b* deficiency impairs IVIg-induced Treg expansion by over 60%, suggesting FcγRIIB participates in antigen-presenting cell tolerogenic programming beyond phagocytosis regulation. Temporally, IVIg injection increases FcγRIIB mRNA levels within 6 hours, peaks at 12 hours, and persists for over 72 hours. This upregulation is particularly significant in antigen-presenting cells of the spleen and liver but weaker in bone marrow-derived macrophages, reflecting tissue microenvironment differences in IVIg responsiveness. Spatially, immunofluorescence shows FcγRIIB+ cells accumulate at the junction of splenic marginal zones and T cell zones, coinciding with the anatomical location of Treg induction.
In conditional knockout studies, DC-specific FcγRIIB deficiency impaired IVIg-induced Treg expansion by over 60%, establishing dendritic cells as critical mediators of IVIg's long-term tolerance effects beyond macrophage phagocytosis.
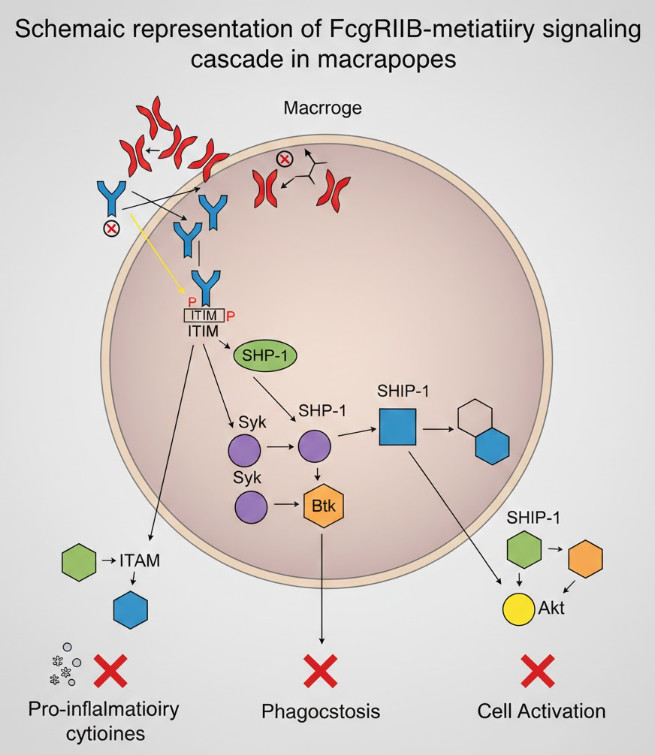
Fig 3. Schematic representation of FcγRIIB-mediated inhibitory signaling cascade in macrophages
3. Fc-Independent Mechanisms: Sialylation-Mediated Regulation of Anti-Inflammatory Effects and Lectin Pathway Analysis
3.1 Preparation and Structural Characteristics of Sialylated IVIg (sIVIg)
The degree of terminal sialylation on the N-297 glycan of IgG Fc is a critical structural determinant of IVIg's anti-inflammatory activity. While standard IVIg preparations contain only ~10-15% IgG molecules with α2,6-linked sialic acid, enzymatic removal of non-sialylated glycans or glycoengineering yields highly sialylated IVIg (containing 60-80% α2,6-sialic acid) that demonstrates significantly enhanced efficacy in arthritis and EAE models at reduced effective doses of 0.2-0.5 g/kg—just one-fifth of standard doses. Mass spectrometry reveals sIVIg glycans are more complex, containing more diantennary or multiantennary structures that enhance affinity for specific lectins. Notably, sIVIg's anti-inflammatory activity is highly sensitive to glycan integrity: neuraminidase treatment removing sialic acid abolishes >90% of its EAE therapeutic effect, while preserving sialic acid but destroying the glycan core also reduces activity by 70%, underscoring the importance of spatial conformation.
3.2 SIGN-R1/DC-SIGN Pathway Signal Transduction Mechanisms
Highly sialylated Fc regions specifically bind C-type lectin SIGN-R1 (mouse homologue) or DC-SIGN (human) on macrophage surfaces, independent of FcγRs, representing a separate anti-inflammatory pathway. This interaction activates intracellular Ca²⁺ signals, activating NF-κB p50 homodimers via SYK and PKCδ to induce *Folr2* (folate receptor 2) expression, thereby promoting STAT3 phosphorylation and sustained IL-10 secretion. In SIGN-R1-deficient mice, sIVIg's therapeutic effect in EAE is lost by >70%, confirming the indispensability of this pathway. Further conditional knockout studies reveal SIGN-R1 is primarily expressed on splenic marginal zone macrophages and specific DC subsets (CD8α⁻CD11b⁺), which display unique transcriptional profiles after sIVIg treatment, including upregulated *Il10*, *Tgfb1*, *Mrc1* (CD206) and downregulated *Tnf*, *Il6*, *Il12b*, forming an M2/tolerogenic phenotype.
3.3 In Vivo Kinetic Differences and Metabolic Fate
Compared to standard IVIg, sIVIg shows 5-fold enhanced accumulation in splenic marginal zone macrophages but a ~30% shorter serum half-life (from 21 to 14 days), suggesting rapid clearance may correlate with enhanced bioactivity. In vivo imaging shows sIVIg generates a significant signal peak in the spleen within 2 hours post-injection, whereas standard IVIg shows more diffuse signal distribution. This difference suggests sIVIg may concentrate its effects on the spleen—a key immunoregulatory organ—through lectin-mediated rapid endocytosis and degradation. Furthermore, sIVIg treatment elevates serum IL-10 levels that peak at 6 hours and remain high at 24 hours, while TNF-α and IL-17A levels continue to decline, creating a favorable anti-inflammatory cytokine milieu. Notably, sIVIg's inhibition of IFN-γ is relatively weak, which may explain its limited efficacy in some Th1-dominant diseases.
High-sialylated IVIg concentrates 5-fold more in splenic marginal zone macrophages despite 30% shorter serum half-life, demonstrating organ-specific targeting via lectin-mediated pathways.
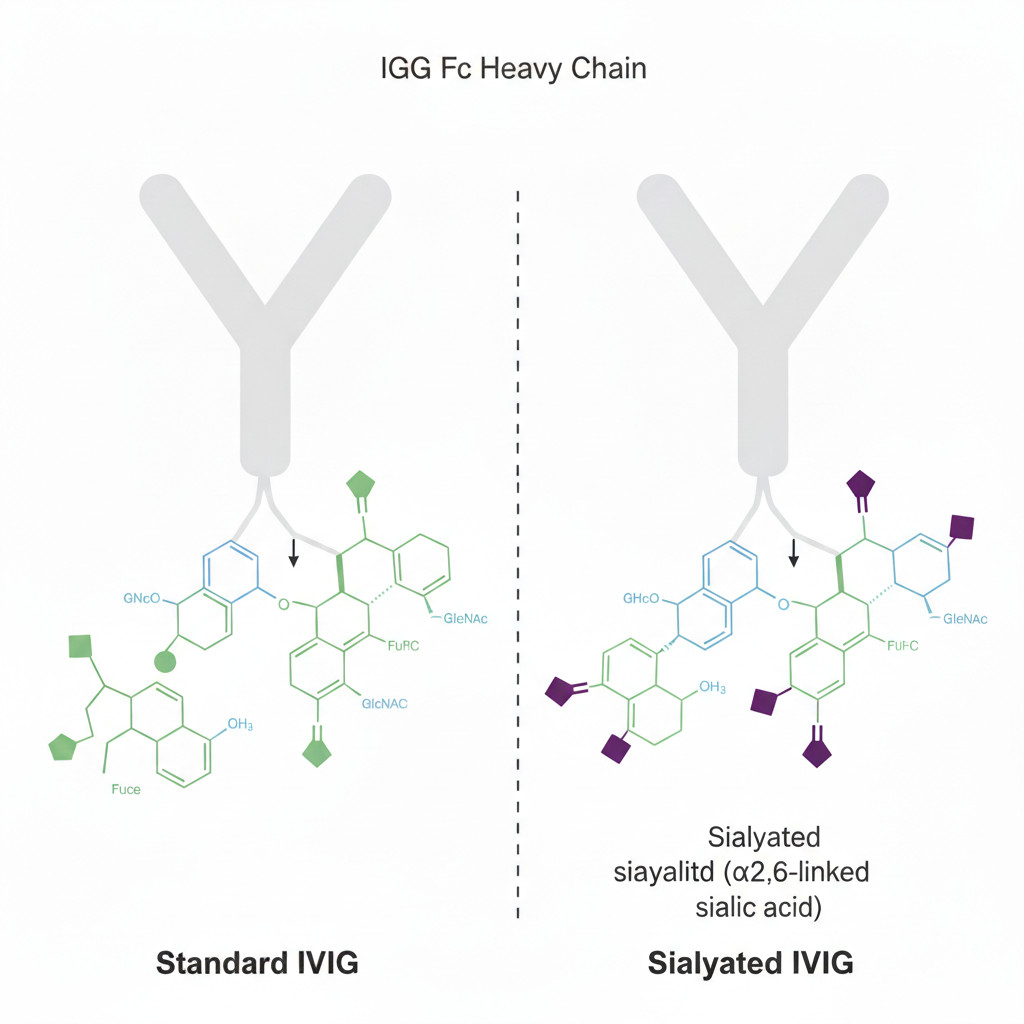
Fig 4: Structural comparison of standard IVIg vs. sialylated IVIg Fc glycans
4. Model Case Studies: Key Data and Mechanistic Integration from EAE and SLE Mouse Models
4.1 Multi-Dimensional Analysis of Experimental Autoimmune Encephalomyelitis (EAE) Model
Model Establishment and Intervention Strategy: In MOG₃₅₋₅₅-induced C57BL/6 mouse EAE model, IVIg (1 g/kg) or highly sialylated IVIg (0.3 g/kg) was administered on days 7 and 14 post-immunization, simulating the clinical therapeutic window. This model recapitulates the CD4⁺ T cell-mediated demyelination pathology of multiple sclerosis (MS).
Clinical and Pathological Improvements:
- Dynamic Clinical Scores: Standard IVIg reduced peak disease scores from 3.5±0.4 to 1.8±0.5 (*p*<0.001), while highly sialylated IVIg further reduced scores to 1.2±0.3 (*p*<0.0001). Notably, early sIVIg intervention (day 3 post-immunization) prevented disease onset, whereas delayed treatment (after day 14) showed diminished efficacy, highlighting the importance of therapeutic windows.
- Histopathology: Spinal cord histology revealed 60% fewer inflammatory foci (H&E staining), 55% reduced CD3⁺ T cell infiltration, and 45% decreased demyelinated area (Luxol fast blue staining) in the IVIg group. The sIVIg group showed 30% higher axonal preservation, suggesting stronger neuroprotective effects.
- Blood-Brain Barrier (BBB) Integrity: Evans blue leakage assays demonstrated 50% reduced BBB disruption after IVIg treatment, correlating with decreased MMP-9 expression and restored tight junction protein (claudin-5, occludin) expression.
Immune Cell Profiles and Molecular Mechanisms:
- T Cell Subset Reprogramming: Flow cytometry confirmed 50-65% fewer CD4⁺IL-17⁺ cells infiltrating the CNS, while Foxp3⁺ Treg proportions increased 2.5-3 fold. In spleen, the Th17/Treg ratio decreased from 2.8 to 0.9, indicating systemic immune environment transformation.
- Antigen-Presenting Cell Function: Splenic DCs showed 2-fold upregulated FcγRIIB expression concomitant with 30-40% decreased CD80/CD86 expression and 70% reduced IL-12p70 secretion. These tolerogenic DCs induced Tregs expressing high levels of LAP (latency-associated peptide) and GARP with enhanced suppressive function.
- Cytokine Microenvironment: Spinal cord homogenates showed 60-70% decreased IL-17A and GM-CSF levels with 3-4 fold increased IL-10 and IL-27p28 after IVIg treatment. This shift was more pronounced in the sIVIg group, where IL-10 levels reached twice those of standard IVIg.
- Transcription Factor Regulation: T cells exhibited decreased RORγt expression with enhanced Foxp3 and STAT5 phosphorylation, suggesting direct regulation of lineage-determining factors by IVIg.
4.2 Long-Term Effect Studies in Systemic Lupus Erythematosus (SLE) Mouse Model
Disease Characteristics and Intervention Protocol of MRL/*lpr* Mice: MRL/*lpr* mice spontaneously produce anti-nuclear antibodies, form immune complexes, and develop lupus nephritis and vasculitis, representing a classic SLE model. In 20-week-old diseased mice, IVIg (1.5 g/kg) was administered weekly for 8 weeks, mimicking chronic disease maintenance therapy.
Autoantibody and Immune Complex Regulation:
- Specific Autoantibodies: Anti-dsDNA antibody titers decreased 55% from baseline 1:1280 to 1:580 (ELISA), while anti-Sm antibodies dropped 40%. Critically, IgG subclass analysis revealed particularly significant decreases (60-70%) in pathogenic IgG2a and IgG2c subclasses, while total IgG levels remained stable, suggesting IVIg selectively inhibits autoreactive B cells.
- Immune Complex Clearance: Circulating immune complexes (CICs) decreased 50%, glomerular IgG deposition reduced 65% (immunofluorescence), and C3 deposition decreased 70%. Hepatic Kupffer cell FcγRIIB-dependent phagocytic function was enhanced, with 2.5-fold upregulated FcγRIIB expression and increased scavenger receptor expression (MARCO, SR-A), promoting CIC clearance and degradation.
Organ Protection and Functional Improvement:
- Renal Function: Proteinuria decreased from baseline 8.2±2.1 mg/24h to 3.1±1.2 mg/24h (*p*<0.001), with 40% improved serum creatinine levels. Renal pathology showed alleviated mesangial proliferation, 50% reduced crescent formation, and decreased interstitial fibrosis.
- Skin and Joint Lesions: Skin ulcer incidence decreased from 60% to 25%, and joint swelling scores dropped 50%, correlating with improved vasculitis and reduced circulating immune complexes.
- Survival Rate: Long-term follow-up revealed IVIg treatment increased 40-week survival from 45% in controls to 75%, extending median survival by 12 weeks.
In-Depth Immunological Mechanism Analysis:
- B Cell Regulation: Splenic B cells showed 50-70% downregulated expression of TLR7/9 signaling pathway genes (*Myd88*, *Irak4*, *Traf6*), suggesting IVIg inhibits nucleic acid-mediated B cell activation. Flow analysis revealed 40% fewer plasma cells (CD138⁺) with increased transitional B cells (T2), implying B cell differentiation blockade.
- T Cell Modulation: Tfh cells (CD4⁺CXCR5⁺PD-1⁺) decreased 45%, while Tfr cells (Treg CXCR5⁺) increased 60%, reducing Tfh/Tfr ratios and suppressing germinal center reactions. Additionally, IVIg-induced Tregs expressed high levels of CTLA-4 and IL-10 with enhanced suppressive capacity.
- Metabolism and Epigenetics: Recent research demonstrates IVIg treatment reduces B cell mitochondrial respiratory function, shifting glycolysis toward oxidative phosphorylation with decreased mTORC1 activity and AMPK activation. This metabolic reprogramming correlates with increased H3K27me3 modification at the *Aicda* (activation-induced cytidine deaminase) gene promoter, suppressing class switch recombination.
Synergistic Action of FcγRIIB and Sialylation Pathways: In SLE models, standard IVIg efficacy partially depends on FcγRIIB (contributing ~60%), whereas highly sialylated IVIg can function independently via the SIGN-R1 pathway, maintaining therapeutic efficacy even in aged mice with impaired FcγRIIB function. This pathway redundancy provides a mechanistic explanation for IVIg's broad clinical effectiveness.
In SLE models, sialylated IVIg maintains therapeutic efficacy in aged mice with impaired FcγRIIB function, demonstrating pathway redundancy that underpins IVIg's broad clinical applicability.
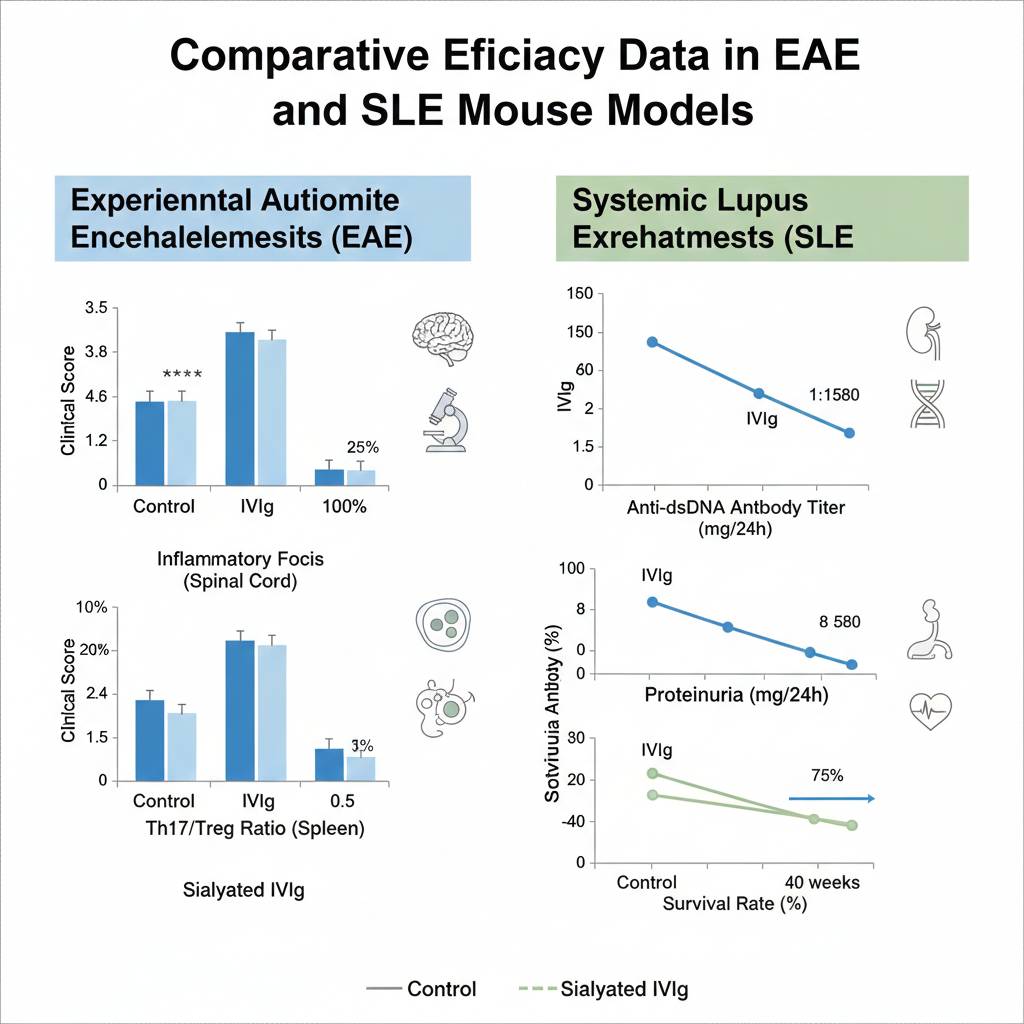
Fig 5. Comparative efficacy data in EAE and SLE mouse models
5. Conclusions and Future Directions: Precision Medicine Prospects for Engineered IVIg
5.1 Summary of Mechanistic Hierarchy and Synergy
IVIg's mechanisms in autoimmune disease animal models exhibit significant hierarchy, synergy, and redundancy. The FcγRIIB-dependent pathway forms the basis for rapid inhibition of effector cell functions, dominating in antibody-mediated diseases like ITP. In contrast, the sialylation-mediated Fc-independent pathway drives durable immune tolerance reprogramming, playing more prominent roles in T cell-mediated diseases like EAE. These two pathways complement each other in complex diseases such as SLE, jointly regulating aberrant activation of B cells, T cells, and innate immune cells.
5.2 Next-Generation Engineered IVIg Development Strategies
Based on deepened mechanistic understanding, future IVIg optimization directions include:
- Highly Sialylated IVIg: Produced via enzymatic modification or glycoengineered cell lines to enhance SIGN-R1/DC-SIGN pathway activation, applicable for T cell-mediated diseases.
- FcγRIIB-Specific Agonists: Engineered Fc regions with enhanced selectivity for inhibitory receptors to reduce high-dose requirements and side effects.
- Bispecific IgG Molecules: Simultaneously targeting pathogenic antigens and FcγRIIB for precise inhibition.
- Metabolic Reprogramming-Enhanced Formulations: Combined with mTOR inhibitors or AMPK agonists to enhance immune tolerance induction.
5.3 Personalized Therapy Potential
For clinical translation, selecting IVIg types based on patient genotypes (such as *FCGR2B* polymorphisms) and disease stage will be key to achieving personalized therapy. For example, patients carrying low-expression promoter variants of *FCGR2B* may benefit more from highly sialylated IVIg, while younger ITP patients may derive greater benefit from standard IVIg.
For precision immunomodulation, selecting IVIg products tailored to disease mechanisms is both a technical decision and a cornerstone of scientific rigor. We advocate for mechanism-based IVIg selection to advance autoimmune disease research and therapy.
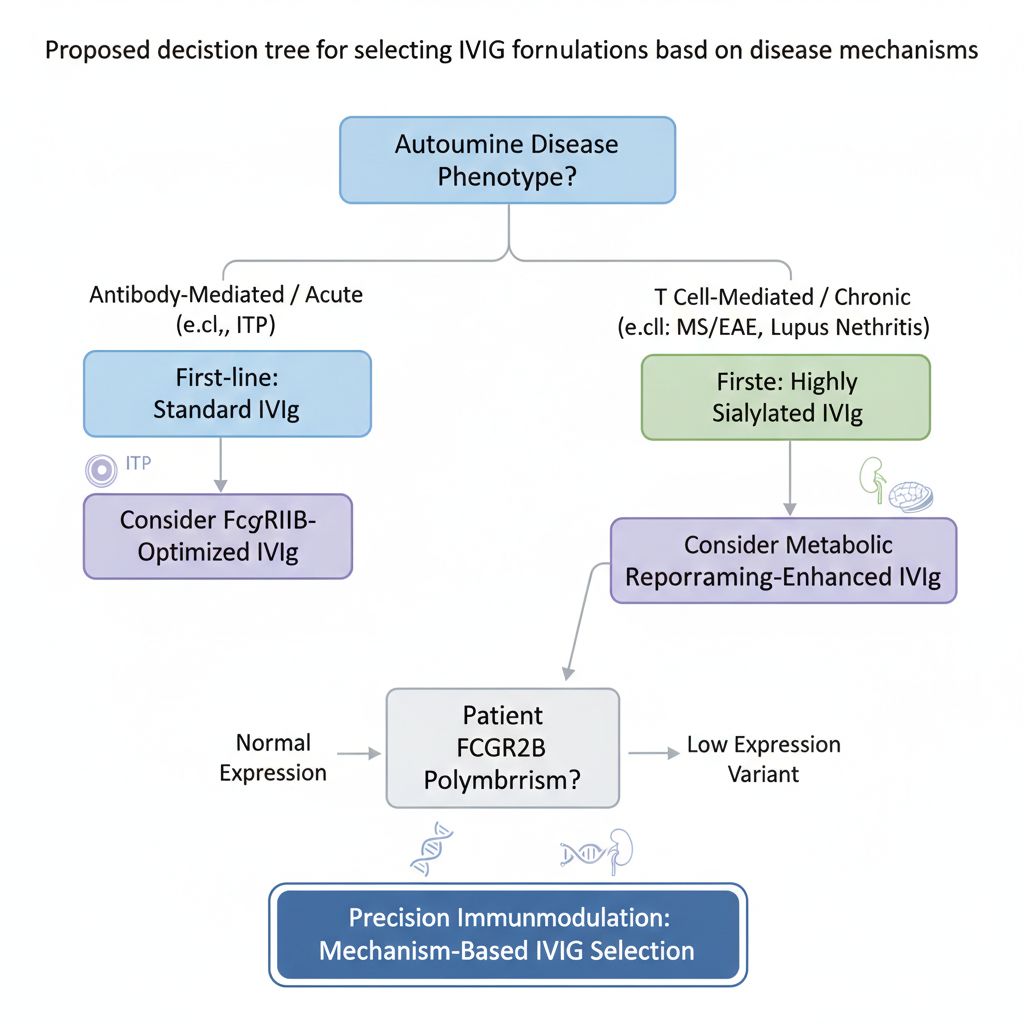
Fig 6. Proposed decision tree for selecting IVIg formulations based on disease mechanisms
References
1. Nimmerjahn, F., & Ravetch, J. V. (2005). Science, 310(5753): 1510-1512.
2. Anthony, R. M., et al. (2008). Science, 320(5874): 373-376.
3. Schwab, I., & Nimmerjahn, F. (2013). Nat Rev Immunol, 13(3): 176-189.
4. Baerenwaldt, A., et al. (2011). J Exp Med, 208(2): 359-368.
5. Siragam, V., et al. (2006). Blood, 107(3): 1075-1080.
6. Crow, A. R., & Lazarus, A. H. (2008). Blood, 111(3): 1479-1480.
7. Kaneko, Y., et al. (2006). Proc Natl Acad Sci USA, 103(52): 19784-19789.
8. De Groot, A. S., et al. (2008). Clin Exp Immunol, 153(3): 333-340.
9. Fiebiger, B. M., et al. (2015). J Allergy Clin Immunol, 135(5): 1241-1253.
10. Mahévas, M., et al. (2020). Nat Rev Immunol, 20(8): 499-510.