
Cat# : THP-0146
| Catalog# | Product Name | Availability | Size | Price | Qty |
|---|---|---|---|---|---|
| THP-0146 | Alglucosidase alfa, Recombinant Human enzyme acid alpha-glucosidase (GAA) | July 29, 2026 | 10ug | $298.00 |
|
| 1mg | $3,998.00 |
|
| Add to Cart Online Order Request a Bulk Order |
| Cat#: | THP-0146 |
| Product Name: |
Alglucosidase alfa, Recombinant Human enzyme acid alpha-glucosidase (GAA)
|
| Cas No: | 420784-05-0 |
| Description: | The product consists of the human enzyme acid alpha-glucosidase (GAA) which is essential for the degradation of glygogen to glucose in lysosomes. It is encoded by the most predominant of nine observed haplotypes of this gene. The product is produced by recombinant DNA technology in a Chinese hamster ovary cell line. The product degrades glycogen by catalyzing the hydrolysis of a-1,4- and a-1,6- glycosidic linkages of lysosomal glycogen. Structurally, the product is a glycoprotein with a calculated mass of 98,008 daltons for the 883 residue mature polypeptide chain, and a total mass of approximately 109,000 daltons, including carbohydrates. |
| Sequences: | AHPGRPRAVPTQCDVPPNSRFDCAPDKAITQEQCEARGCCYIPAKQGLQGAQMGQPWCFFPPSYPSYKLENLSSSEMGYTATLTRTTPTFFPKDILTLRLDVMMETENRLHFTIKDPANRRYEVPLETPHVHSRAPSPLYSVEFSEEPFGVIVRRQLDGRVLLNTTVAPLFFADQFLQLSTSLPSQYITGLAEHLSPLMLSTSWTRITLWNRDLAPTPGANLYGSHPFYLALEDGGSAHGVFLLNSNAMDVVLQPSPALSWRSTGGILDVYIFLGPEPKSVVQQYLDVVGYPFMPPYWGLGFHLCRWGYSSTAITRQVVENMTRAHFPLDVQWNDLDYMDSRRDFTFNKDGFRDFPAMVQELHQGGRRYMMIVDPAISSSGPAGSYRPYDEGLRRGVFITNETGQPLIGKVWPGSTAFPDFTNPTALAWWEDMVAEFHDQVPFDGMWIDMNEPSNFIRGSEDGCPNNELENPPYVPGVVGGTLQAATICASSHQFLSTHYNLHNLYGLTEAIASHRALVKARGTRPFVISRSTFAGHGRYAGHWTGDVWSSWEQLASSVPEILQFNLLGVPLVGADVCGFLGNTSEELCVRWTQLGAFYPFMRNHNSLLSLPQEPYSFSEPAQQAMRKALTLRYALLPHLYTLFHQAHVAGETVARPLFLEFPKDSSTWTVDHQLLWGEALLITPVLQAGKAEVTGYFPLGTWYDLQTVPVEALGSLPPPPAAPREPAIHSEGQWVTLPAPLDTINVHLRAGYIIPLQGPGLTTTESRQQPMALAVALTKGGEARGELFWDDGESLEVLERGAYTQVIFLARNNTIVNELVRVTSEGAGLQLQKVTVLGVATAPQQVLSNGVPVSNFTYSPDTKVLDICVSLLMGEQFLVSWC |
| Species: | Human |
| Molecular Weight: | 105270.802 Da |
| Source: | CHO |
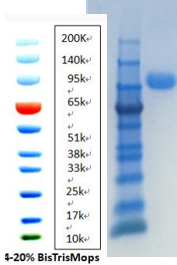
| Purity: | >99% by SDS-Page and HPLC analysis |
| Formula: | C4435H6739N1175O1279S32 |
| Endotoxin: | <0.001 EU per 1 μg by the LAL method |
| Storage: | Lyophilized protein should be stored at < -20°C, though stable at room temperature for 3 weeks. Reconstituted protein solution can be stored at 2-8 °C for 1 week. Aliquots of reconstituted samples are stable at < -20°C for 3 months. |
| Application: | For the treatment of Pompe disease (GAA deficiency) in infants and pediatric patients. |
| Concentration: | 5 mg/1mL |
| Publication: |
|
|
|
For more information on how our products could help advance your project, please contact us.
ENTER YOUR EMAIL HERE TO SUBSCRIBE.
Copyright © 2026 Creative BioMart. All Rights Reserved.
