| Catalog# | Product Name | Size | Price | Qty | Inquiry |
|---|---|---|---|---|---|
| THP-0313 | Velmanase alfa, Recombinant human alpha mannosidase | 1 vial | $3,998.00 |
|
Add to Cart Order |
Alpha-mannosidosis, also known as a lysosomal alpha-D-mannosidase deficiency, is a rare lysosomal storage disorder with autosomal recessive inheritance characterized by a deficiency of alpha-D-mannosidase. The disease can affect individuals of any race or ethnicity, and its prevalence is estimated at 1 in 500,000 worldwide.
Alpha-mannosidosis is caused by mutations in the MAN2B1 gene that interfere with the ability of alpha-mannosidase to perform its function correctly, resulting in the accumulation of complex sugar molecules in cells and causing organ and tissue damage. Symptoms that may result include mental retardation, bone abnormalities, hearing loss, muscle weakness, coarse facial features, increased susceptibility to infection, and problems controlling body movement.
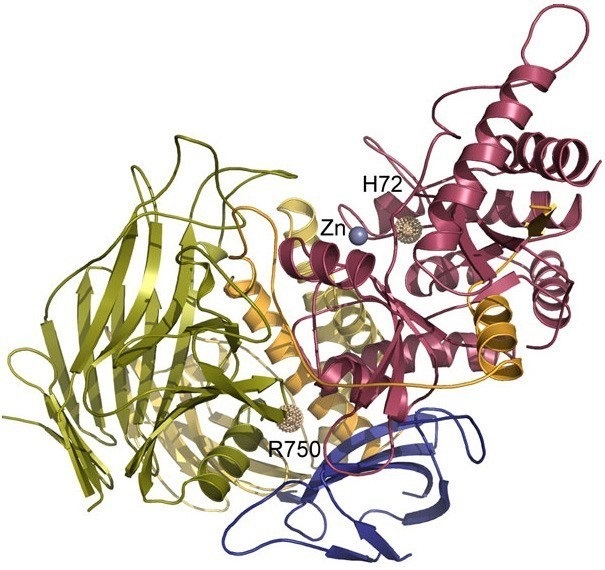
Figure1. The 3-dimensional structure of lysosomal α-mannosidase. (Malm D., et al., 2008)
Alpha-mannosidase is located in the lysosomes of cells and is responsible for the breakdown and recycling of specific complex sugars containing sugar molecules called mannose. More than 2,000 α-mannosidase gene sequences have been sequenced and are found mainly in the endoplasmic reticulum, Golgi apparatus, lysosomes, cytosol, and other organelles of humans, mice, cattle, birds, insects, bacteria, and plants.
Alpha-mannosidases are mainly involved in protein glycosylation and glycoprotein glycan hydrolysis modifications. Glycosylation and glycoprotein glycan hydrolysis are closely related to the folding, maturation, sorting, transport, conformational maintenance, half-life, and biological activity of nascent glycoproteins, which in turn play a role in cell adhesion, inflammatory response, hormonal activity, arthritis, immune surveillance and cancer cell metastasis.
Symptoms of α-mannosidosis range from mild to moderate and severe, with individuals with the early-onset severe and the rapidly progressive disease typically not surviving beyond childhood, while those with mild disease can survive into adult life. Although there is no fundamental treatment for α-mannosidosis, regulation of α-mannosidase may provide a new way to treat related diseases including α-mannosidosis, congenital dyserythropoietic anemia type II, etc.
For example, enzyme replacement therapy with recombinant human alpha-mannosidase is scheduled for clinical development in the United States in 2020 and is already approved in the European Union for the treatment of non-neurological manifestations of mild to moderate disease. Therefore Creative BioMart offers recombinant human alpha-mannosidase products to assist researchers to better conduct various studies in alpha-mannosidosis.
As a leading supplier of recombinant proteins and related products, Creative BioMart takes pride in helping researchers solve real-world problems, just as we look forward to the full application of recombinant human alpha-mannosidase in the treatment of alpha-mannosidosis. If you are conducting or planning to conduct research related to α-mannosidosis and need recombinant human alpha-mannosidase products, then we are the company to call. You can contact our staff directly or submit your product request online and we will contact you as soon as possible to discuss the details of your order.
References
For more information on how our products could help advance your project, please contact us.
ENTER YOUR EMAIL HERE TO SUBSCRIBE.
Copyright © 2026 Creative BioMart. All Rights Reserved.